chevron_leftchevron_right

We have only begun to scratch the surface in understanding mammalian development. An overwhelming caveat is the lack of topographical transcriptomic information to correlate signaling cues and cell-cell interactions within the hierarchy of cell fate decisions. Spatially resolved transcriptomic technologies are promising tools to fill this gap. However, the small field of view and imbalance between resolution and transcript capture of current methodologies precludes their systematic application to study relatively large and multilayered embryos. Here, we have combined DNA nanoball (DNB) patterned arrays and in situ RNA capture to create SpaTial Enhanced REsolution Omics-sequencing (Stereo-seq). This approach allows transcriptomic profiling of large histological sections with high resolution, sensitivity, and reproducibility. We have applied Stereo-seq to study the kinetics of transcriptional variation, the networks of transcription factor binding events and their relationship with morphogens across spatial domains of gene expression in a time course of mouse organogenesis. We have used this information to detect the emergence of tissue-specific cell identities such as early neuroblast populations in the late neural tube stage or the gradients of neuronal specification in the neocortex. Furthermore, we have mapped the expression of a panel of developmental disease-related loci to define the spatiotemporal windows of tissue vulnerability. Our panoramic atlas constitutes a essential resource to investigate longstanding questions concerning the molecular basis of normal and abnormal mammalian development.

Recent discoveries about the molecular heterogeneity of the cerebellar cortex suggest the existence of functionally divergent subclasses of anatomically defined cell types. Using spatial transcriptome and single-nucleus RNA-seq analysis, we mapped 3D transcriptomic atlases of the whole cerebellum of mice, marmosets, and macaques at the single-cell resolution. Comparative analysis revealed specific cell types, cell localizations, and intra-cerebellum molecular heterogeneity across species. A comprehensive database generated from this study will expand the acknowledgment of the mammalian cerebellum.
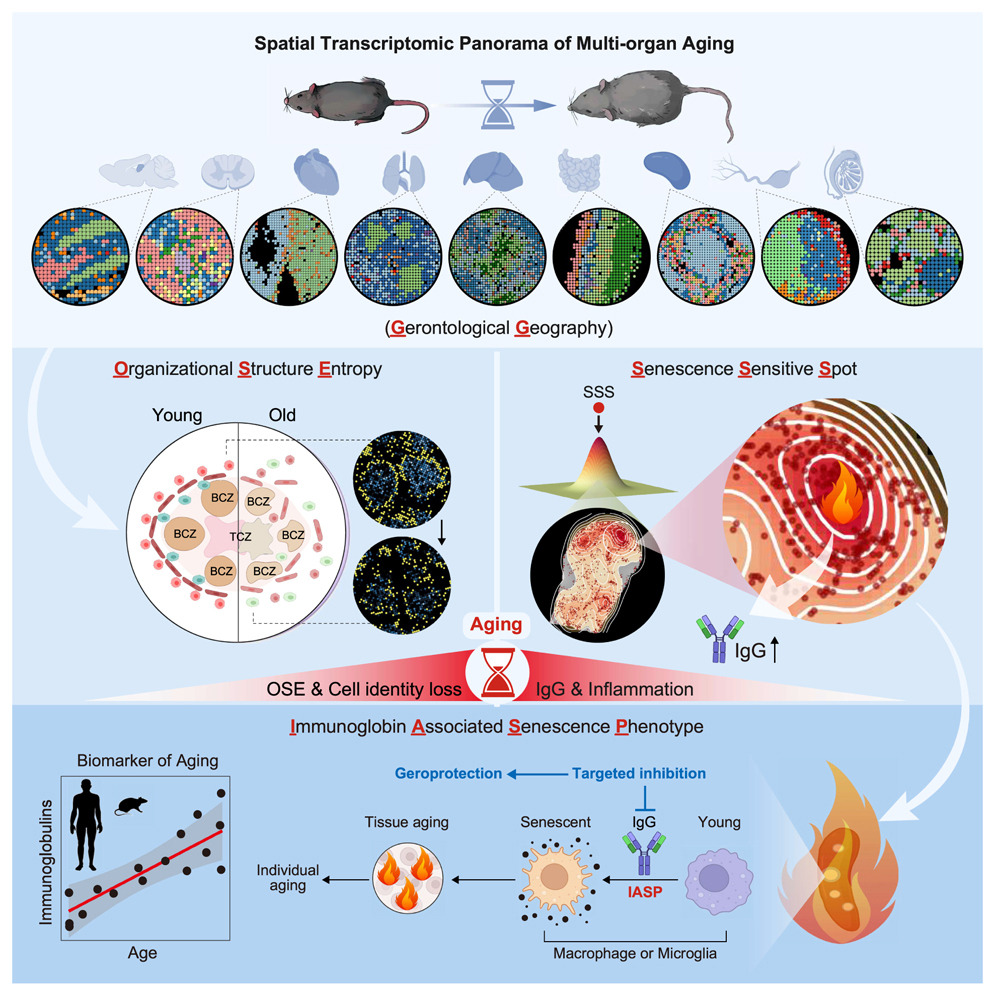
3Spatial transcriptomic landscape unveils immunoglobin-associated senescence as a hallmark of aging
(ID: STDS0000247)
To systematically characterize the loss of tissue integrity and organ dysfunction resulting from aging, we produced an in-depth spatial transcriptomic profile of nine tissues in male mice during aging. We showed that senescence-sensitive spots (SSSs) colocalized with elevated entropy in organizational structure and that the aggregation of immunoglobulin-expressing cells is a characteristic feature of the microenvironment surrounding SSSs. Immunoglobulin G (IgG) accumulated across the aged tissues in both male and female mice, and a similar phenomenon was observed in human tissues, suggesting the potential of the abnormal elevation of immunoglobulins as an evolutionarily conserved feature in aging. Furthermore, we observed that IgG could induce a pro-senescent state in macrophages and microglia, thereby exacerbating tissue aging, and that targeted reduction of IgG mitigated aging across various tissues in male mice. This study provides a high-resolution spatial depiction of aging and indicates the pivotal role of immunoglobulin-associated senescence during the aging process.
Yuzhe Sun
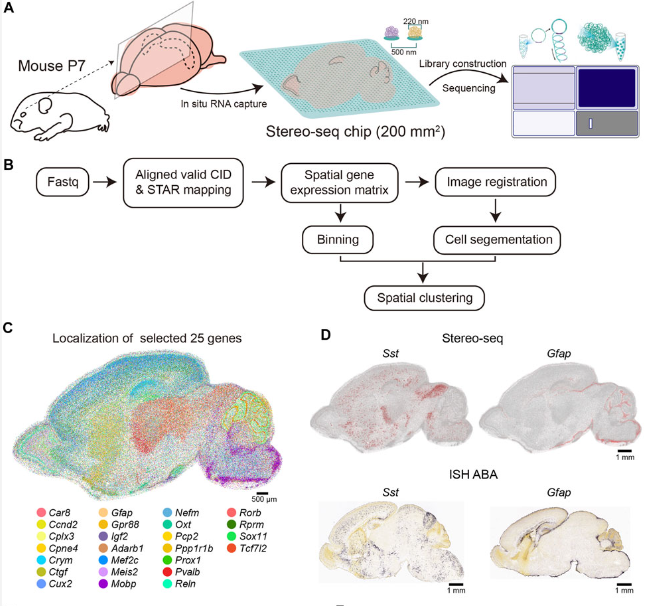
4A cellular resolution spatial transcriptomic landscape of the postnatal mouse brain
(ID: STDS0000139)
Here we apply Stereo-seq to generate a spatially-resolved transcriptomic description of the postnatal day 7 (P7) murine whole brain sagittal section near the middle line. Our study comprehensively dissected the anatomical regions, gene expression and gene regulatory network patterns and cell type localization at whole brain scale.
Mengnan cheng; Liang Wu; Chuanyu Liu; Longqi Liu
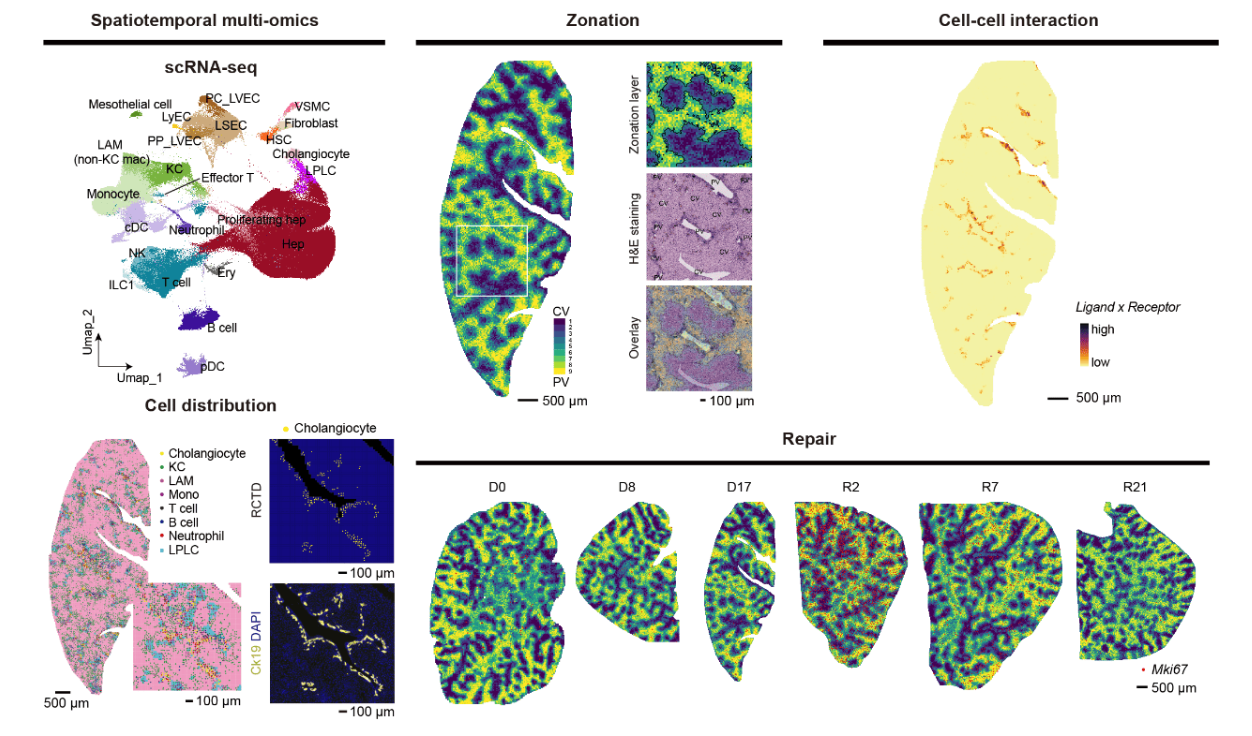
Cholestatic injuries, characterized by regional damage around the periportal region, lack curative therapies and cause considerable mortality. In this study, we generated a high-definition spatiotemporal atlas during cholestatic injury and repair by Stereo-seq and single-cell transcriptomics. We uncovered that cholangiocytes function as a periportal hub (cholangio-hub) by integrating multiple signals with neighboring cells. Feedback between cholangiocytes and lipid-associated macrophages (LAM) was detected in the cholangio-hub, which is related to the differentiation of LAM, a recently identified subpopulation of macrophages crucial in tissue injury. Moreover, the cholangio-hub highly expressed TGFβ, which is associated with cholangiocyte conversion of liver progenitor-like cells during injury and dampened proliferation of periportal hepatocytes during recovery. Importantly, spatiotemporal analysis revealed a key inhibitory rheostat for hepatocyte proliferation. Our data provide a comprehensive resource for demarcating regional cholestatic injuries.
Shijie Hao

6Spatial transcriptomics map of the embryonic mouse brain – a tool to explore neurogenesis
(ID: STDS0000235)
The developing brain has a complex and well-organized anatomical structure comprising different types of neural and non-neural cells. Stem cells, progenitors, and newborn neurons tightly interact with their neighbouring cells and tissue microenvironment, and this intricate interplay ultimately shapes the output of neurogenesis. Given the relevance of spatial cues during brain development, we acknowledge the necessity for a transcriptomics atlas within the tissue context accessible to the neurodevelopmental community. To fulfil this need, we offer an open-access spatial gene expression browser of the embryonic mouse brain at the peak of neurogenesis. Using 10x Visium technology, we generated spatially-resolved RNAseq data from E13.5 embryonic brain sections. Unsupervised clustering reliably defined specific cell type populations of diverse lineages and maturational states. Differential expression analysis revealed unique transcriptional signatures across specific embryonic brain areas, uncovering novel features inherent to particular anatomical domains. Furthermore, we integrated single-cell RNAseq data from E13.5 mouse brains into our Spatial Transcriptomics data, adding tissue context to single-cell resolution. In summary, we provide a valuable tool that enables the exploration and discovery of unforeseen molecular players involved in neurogenesis, particularly in the crosstalk between different cell types.
Di Marco B, Vázquez-Marín J, Monyer H, Centanin L, Alfonso J

7Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment
(ID: STDS0000153)
Transcriptional heterogeneity among malignant cells of a tumor has been studied in individual cancer types and shown to be organized into cancer cell states; however, it remains unclear to what extent these states span tumor types, constituting general features of cancer. Here, we perform a pan-cancer single-cell RNA-Seq analysis across 15 cancer types and identify a catalog of gene modules whose expression defines recurrent cancer cell states including ‘stress’, ‘interferon response’, ‘epithelial-mesenchymal transition’, ‘metal response’, ‘basal’ and ‘ciliated’. Spatial transcriptomic analysis linked the interferon response in cancer cells to T cells and macrophages in the tumor microenvironment. Using mouse models, we further found that induction of the interferon response module varies by tumor location and is diminished upon elimination of lymphocytes. Our work provides a framework for studying how cancer cell states interact with the tumor microenvironment to form organized systems capable of immune evasion, drug resistance, and metastasis.
Barkley, Dalia,Yanai, Itai
8Multiomic researches revealed a spatial-temporal cellular atlas of mouse liver regeneration
(ID: STDS0000059)
Despite the liver is one of the largest and most important organs in mammals, hepatic cell responses in homeostasis and perturbation are yet poorly understood. This is due to the difficulty of systematically studying multiple cell types in different cell states and locations, many of which are transient. Here, we used Stereo-seq (Spatio-Temporal Enhanced REsolution Omics-sequencing) combined with high-throughput single-cell transcriptomic analysis to profile murine liver homeostasis and regeneration after partial resection. Our integrative analysis dissects with unprecedented resolution the transcriptomic gradients controlling liver cell function at whole lobe scale, carefully defining how genes and gene regulatory networks are modulated through intercellular communication. Among other important regulators, we identified the transcriptional cofactor TBL1XR1 as an inflammation-induced master switch derepressing genes necessary for hepatocyte proliferation including metabolic genes and Wnt/β-catenin signaling pathway components. Our works lays the foundation for future high-definition spatiotemporal studies of liver malfunction.
Shijie Hao; Jiangshan Xu; Quan Shi; Pengcheng Guo; Shuncheng Shangguan

Revealing the relationships between the tissue structures and transcriptomic information is significant for understanding the mechanisms and principles underpinning the biological processes in lung development and diseases. The widely transcriptomic technics, including bulk and single-cell RNA sequencing, cannot accurately restore the tissue location of the gene expression. The advanced spatial transcriptomic tools can fill this gap properly with in-situ RNA capturing and spatial barcode labeling and translating. However, the published spatial transcriptome data of lung is rare or low-quality. Here we harnessed the large-view and high-resolution capabilities of Stereo-seq, one of the most advanced spatial transcriptome tools, in a 5-week-old female mouse to uncover the lung functions with their tissue location resolved. We also found a gradient expression of a bunch of genes related to cell proliferation along the axis of the proximal-to-distal trachea. All the data provided by the study pave the way for further studies in lung development and disease in the future.
Yujia Jiang,Shijie Hao

Tumor heterogeneity is a major challenge for oncology drug discovery and development. Understanding of the spatial tumor landscape is key to identifying new targets and impactful model systems. Here, we test the utility of spatial transcriptomics (ST) for Oncology Discovery by profiling 40 tissue sections and 80,024 capture spots across a diverse set of tissue types, sample formats, and RNA capture chemistries. We verify the accuracy and fidelity of ST by leveraging matched pathology analysis that provide a ground truth for tissue section composition. We then use spatial data to demonstrate the capture of key tumor depth features, identifying hypoxia, necrosis, vasculature, and extracellular matrix variation. We also leverage spatial context to identify relative cell type locations showing the anti-correlation of tumor and immune cells in syngeneic cancer models. Lastly, we demonstrate target identification approaches in clinical pancreatic adenocarcinoma samples, highlighting tumor intrinsic biomarkers and paracrine signaling.
Lyubetskaya, Anna,Rabe, Brian,Fisher, Andrew,Lewin, Anne,Neuhaus, Isaac,Brett, Connie,Brett, Todd,Pe