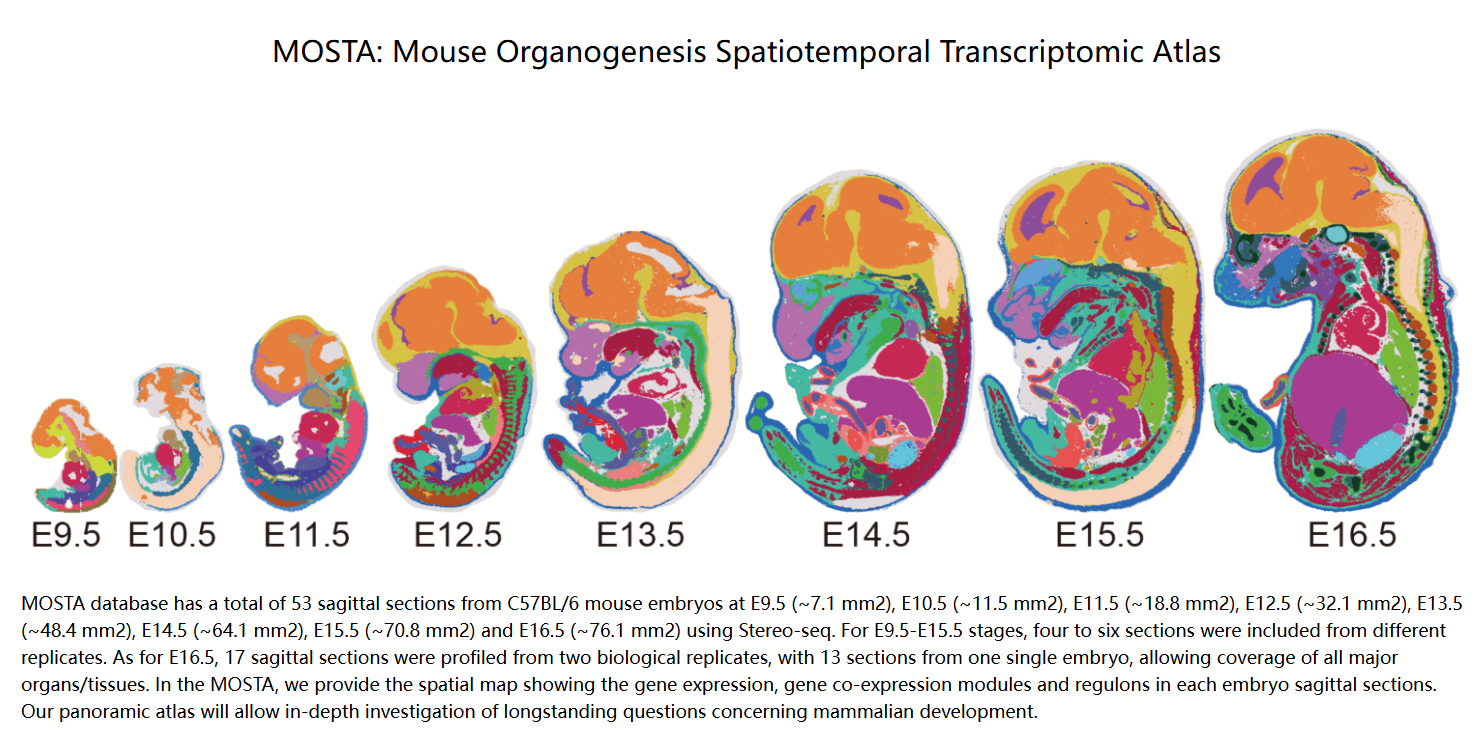
2022-05-04
Chen, A., Liao, S., Cheng, M., Ma, K., Wu, L., Lai, Y., ... & Wang, J. (2022). Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell, 185(10), 1777-1792.
DOI:10.1016/j.cell.2022.04.003
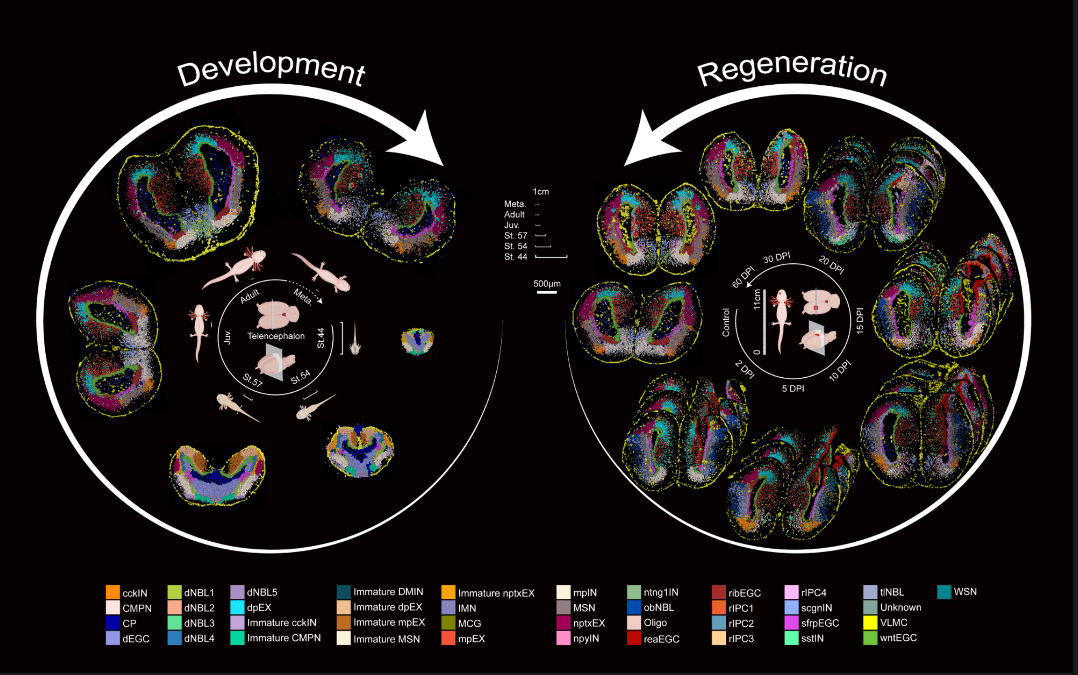
2022-09-02
Wei, X., Fu, S., Li, H., Liu, Y., Wang, S., Feng, W., ... & Gu, Y. (2022). Single-cell Stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science, 377(6610), eabp9444.
DOI:10.1126/science.abp9444
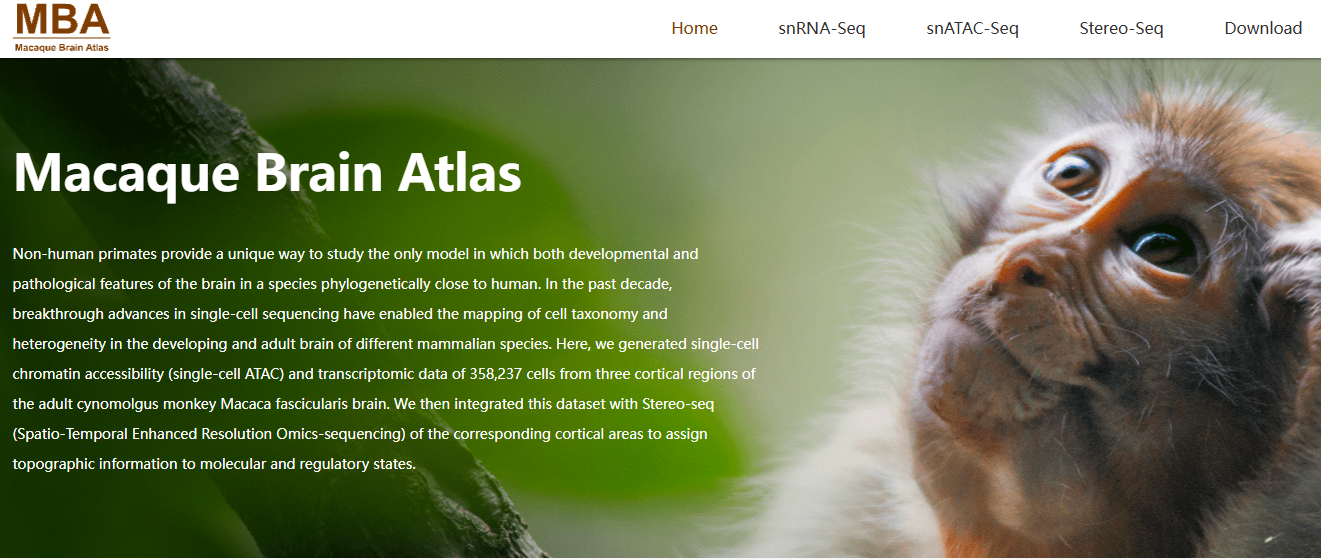
2022-11-08
Lei, Y., Cheng, M., Li, Z., Zhuang, Z., Wu, L., Sun, Y., ... & Xu, X. (2022). Spatially resolved gene regulatory and disease-related vulnerability map of the adult Macaque cortex. Nature Communications, 13(1), 6747.
DOI:10.1038/s41467-022-34413-3
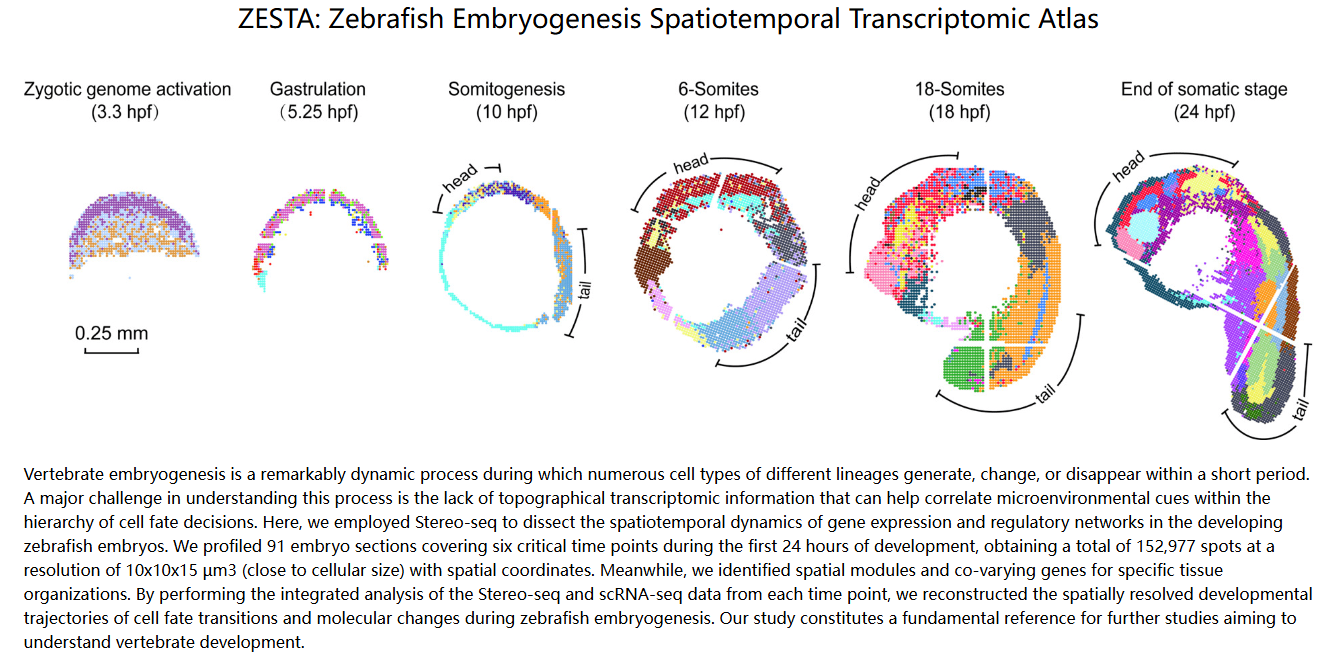
2022-05-04
Liu, C., Li, R., Li, Y., Lin, X., Zhao, K., Liu, Q., ... & Liu, L. (2022). Spatiotemporal mapping of gene expression landscapes and developmental trajectories during zebrafish embryogenesis. Developmental Cell, 57(10), 1284-1298.
DOI:10.1016/j.devcel.2022.04.009
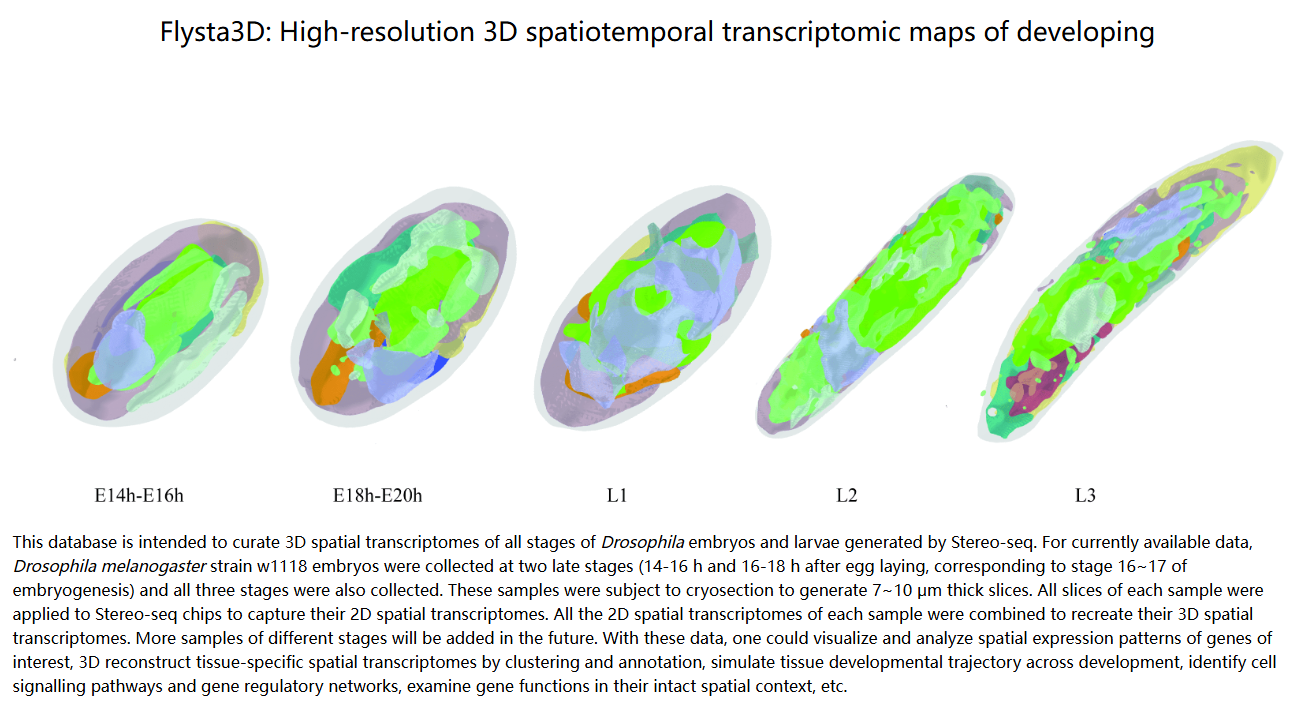
2022-05-04
Wang, M., Hu, Q., Lv, T., Wang, Y., Lan, Q., Xiang, R., ... & Liu, L. (2022). High-resolution 3D spatiotemporal transcriptomic maps of developing Drosophila embryos and larvae. Developmental Cell, 57(10), 1271-1283.
DOI:10.1016/j.devcel.2022.04.006
2022-01-14
Xia, Keke et al. “The single-cell stereo-seq reveals region-specific cell subtypes and transcriptome profiling in Arabidopsis leaves.” Developmental cell vol. 57,10 (2022): 1299-1310.e4.
DOI:10.1016/j.devcel.2022.04.011
2024-02-06
Zhang, Xianglong et al. “A transcriptomic and proteomic atlas of obesity and type 2 diabetes in cynomolgus monkeys.” Cell reports vol. 42,8 (2023): 112952.
DOI:10.1016/j.celrep.2023.112952
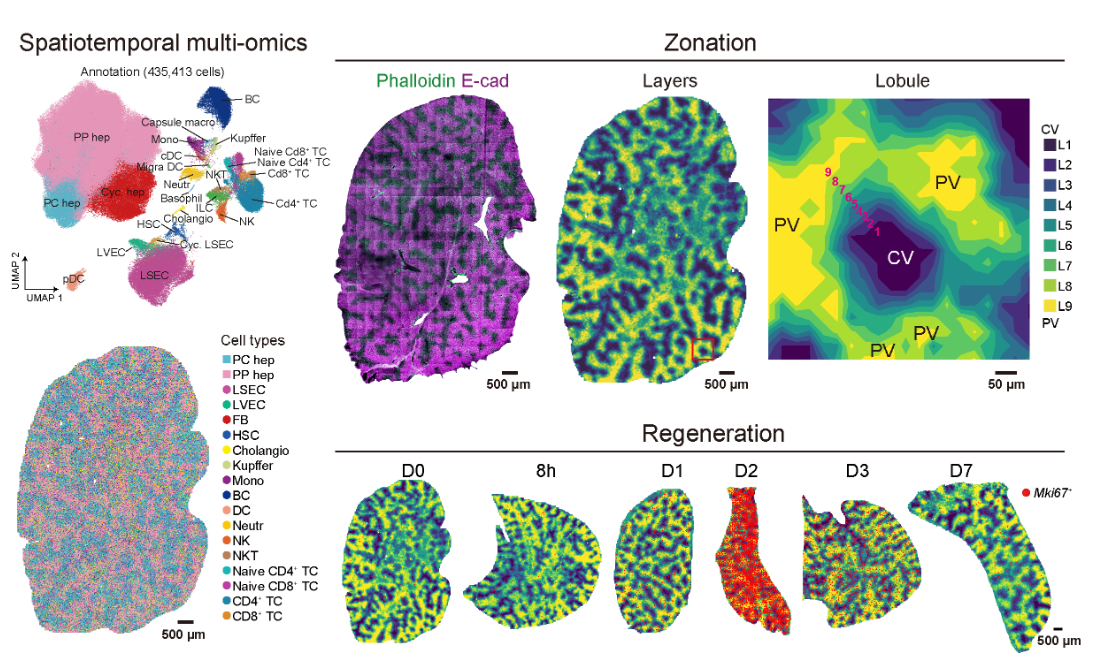
2024-04-16
Xu, J., Guo, P., Hao, S. et al. A spatiotemporal atlas of mouse liver homeostasis and regeneration. Nat Genet (2024).
DOI:10.1038/s41588-024-01709-7
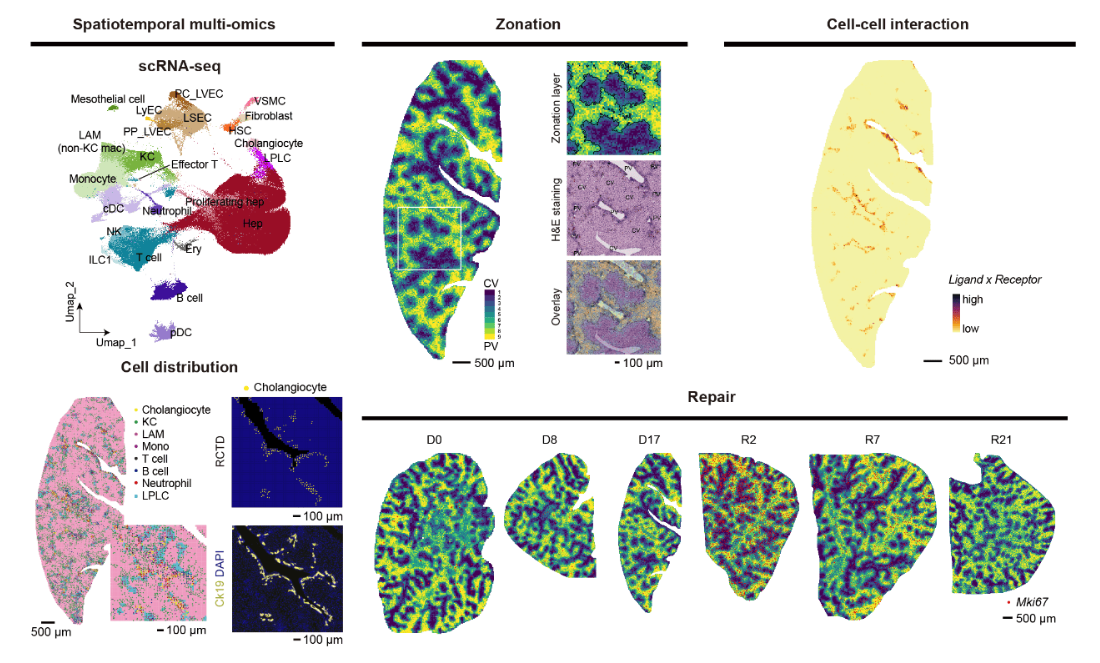
2024-04-16
Wu, B., Shentu, X., Nan, H. et al. A spatiotemporal atlas of cholestatic injury and repair in mice. Nat Genet (2024).
DOI:10.1038/s41588-024-01687-w
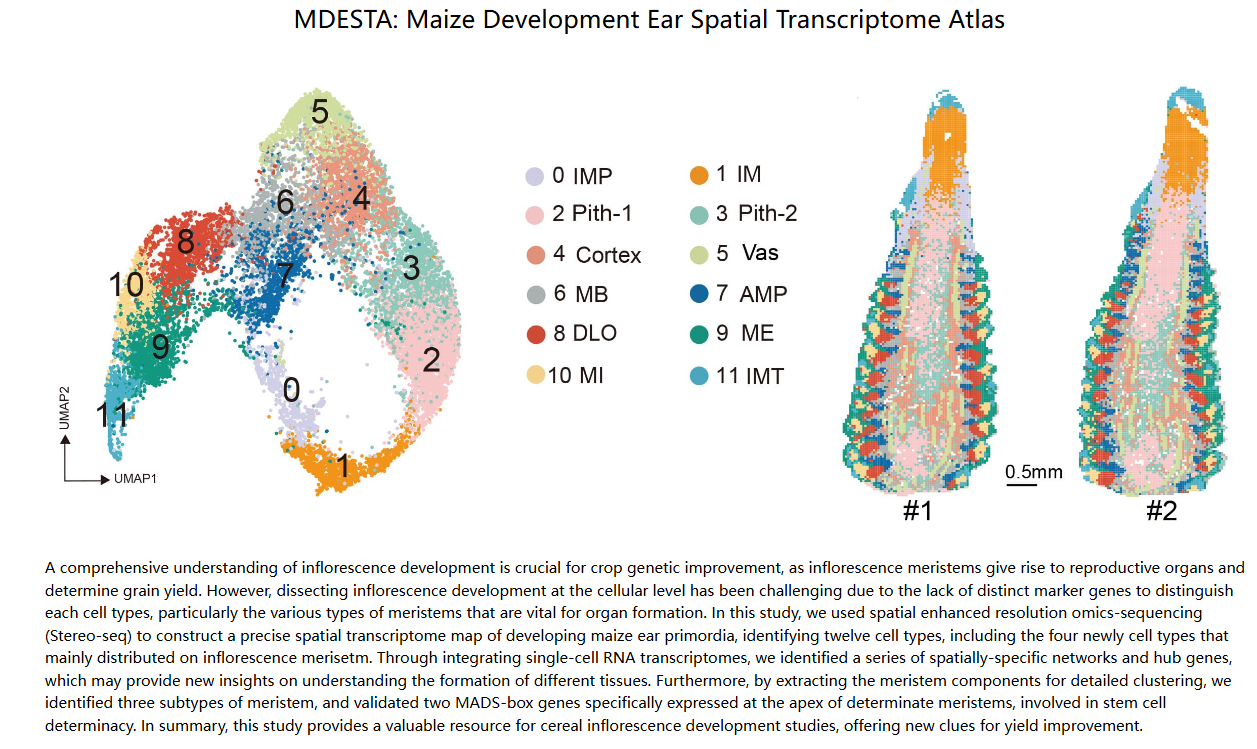
2024-05-14
Wang, Y., Luo, Y., Guo, X. et al. A spatial transcriptome map of the developing maize ear. Nat. Plants (2024).
DOI:10.1038/s41477-024-01683-2

2024-09-27
Shijie Hao et al. ,Cross-species single-cell spatial transcriptomic atlases of the cerebellar cortex.Science385,eado3927(2024).
DOI:10.1126/science.ado3927
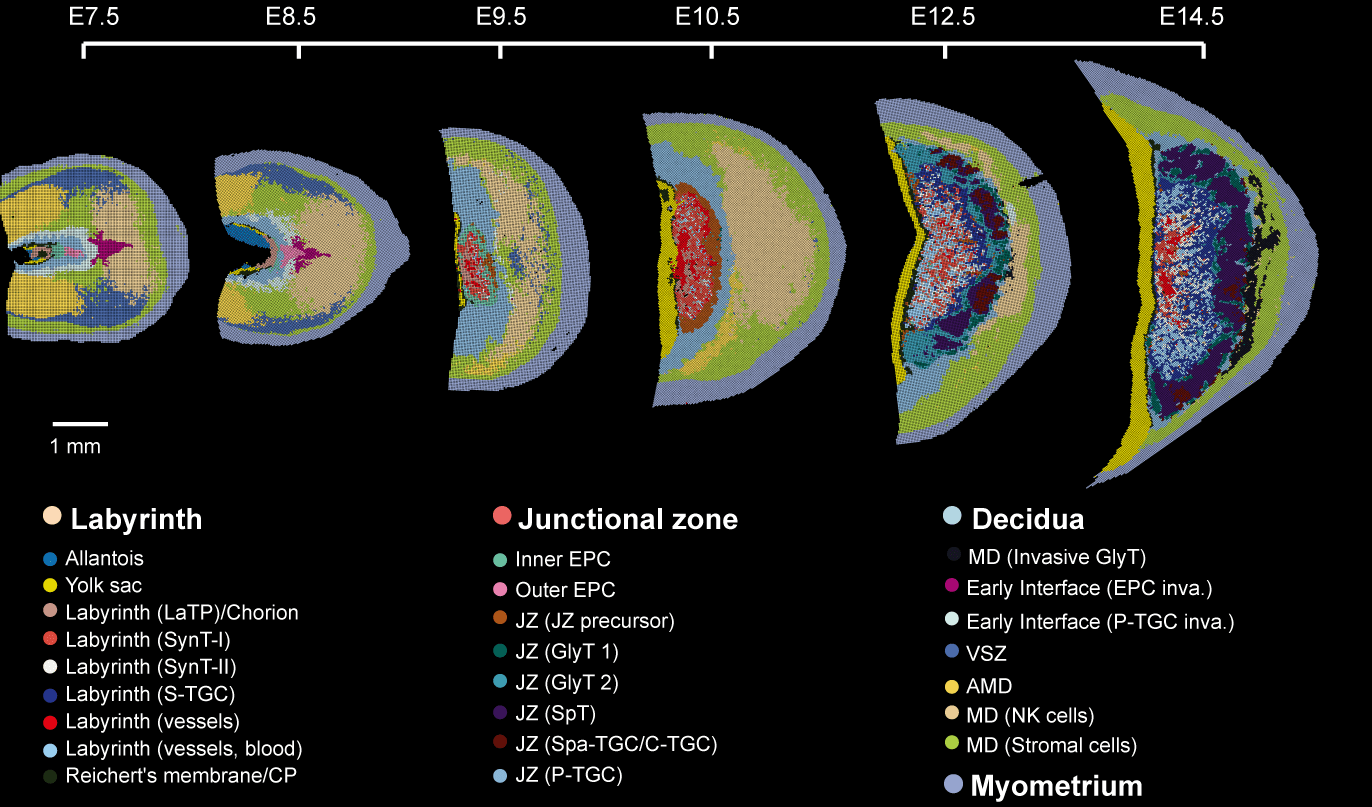
2024-10-22
Wu, Y., Su, K., Zhang, Y. et al. A spatiotemporal transcriptomic atlas of mouse placentation. Cell Discov 10, 110 (2024).
DOI:10.1038/s41421-024-00740-6

2025-02-18
1. Fan, Jingwei et al. “A large-scale integrated transcriptomic atlas for soybean organ development.” Molecular plant, S1674-2052(25)00069-3. 18 Feb.2025.
DOI:10.1016/j.molp.2025.02.003
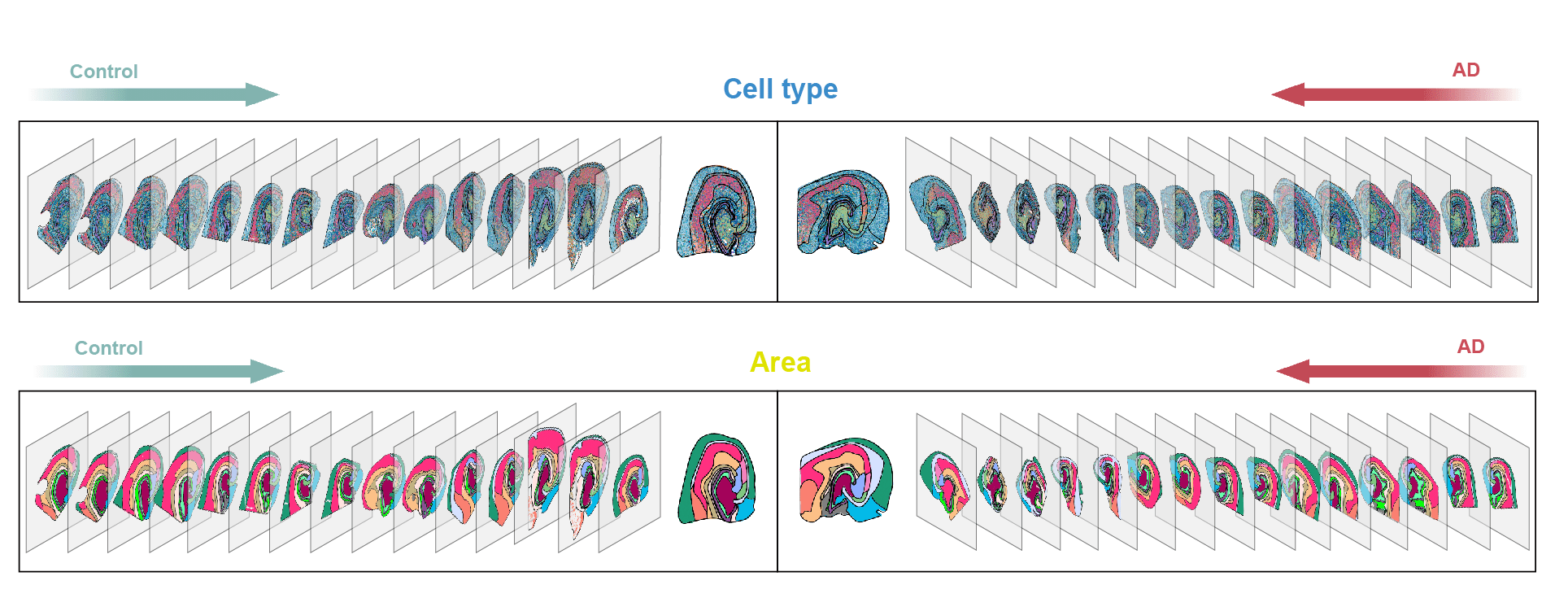
2025-03-31
Wang, P., Han, L., Wang, L., Tao, Q., Guo, Z., Luo, T., ... & Zhang, J. (2025). Molecular Pathways and Diagnosis in Spatially Resolved Alzheimer’s Hippocampal Atlas. Neuron. Advance online publication.
DOI:10.1016/j.neuron.2025.03.002
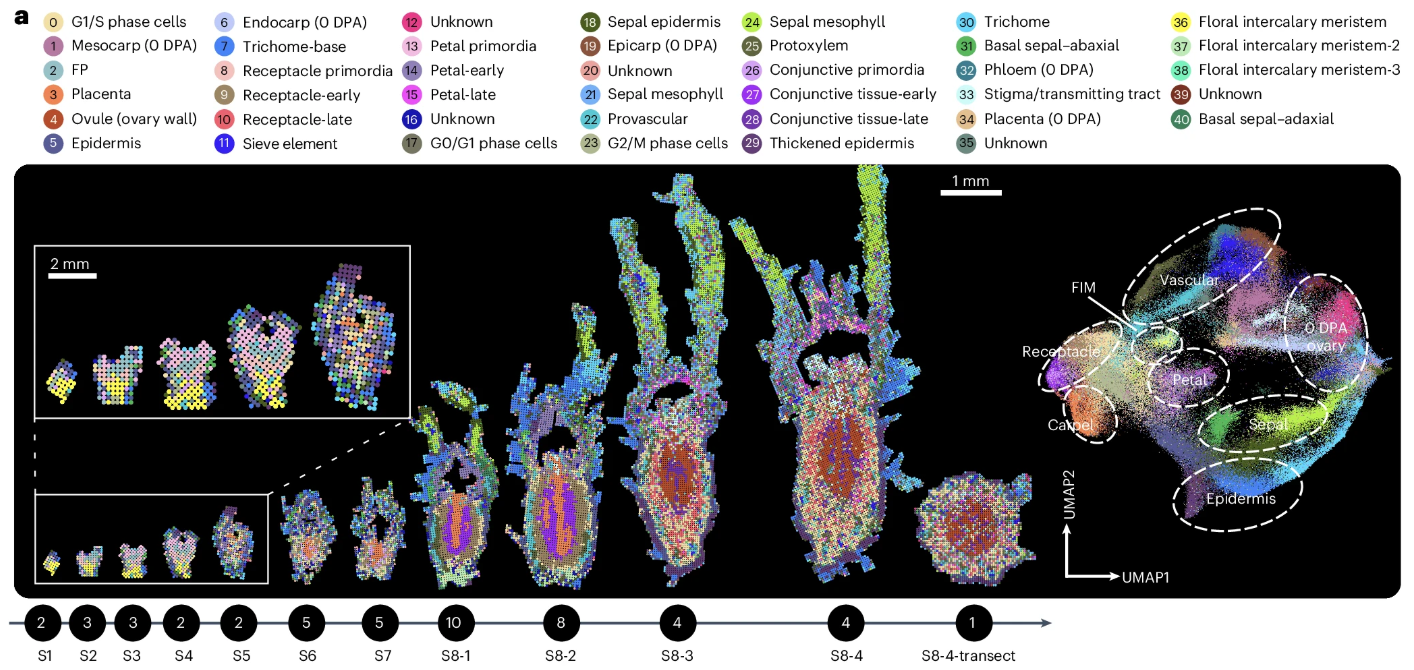
2025-04-01
Dong, Z., Liu, X., Guo, X. et al. Developmental innovation of inferior ovaries and flower sex orchestrated by KNOX1 in cucurbits. Nat. Plants (2025).
DOI:10.1126/science.ado3927
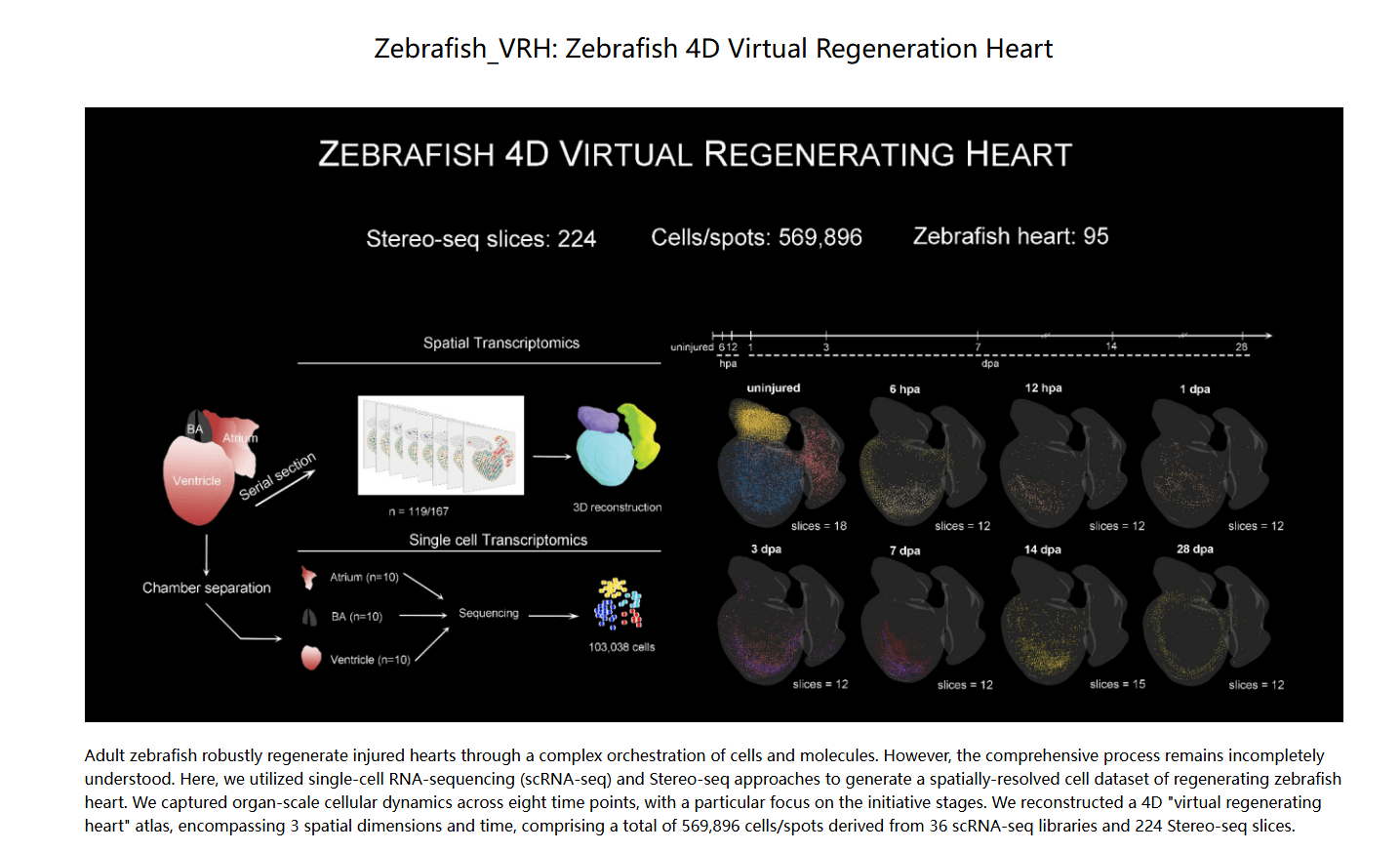
2025-04-19
Li, L., Lu, M., Guo, L. et al. An organ-wide spatiotemporal transcriptomic and cellular atlas of the regenerating zebrafish heart. Nat Commun 16, 3716 (2025).
DOI:10.1038/s41467-025-59070-0
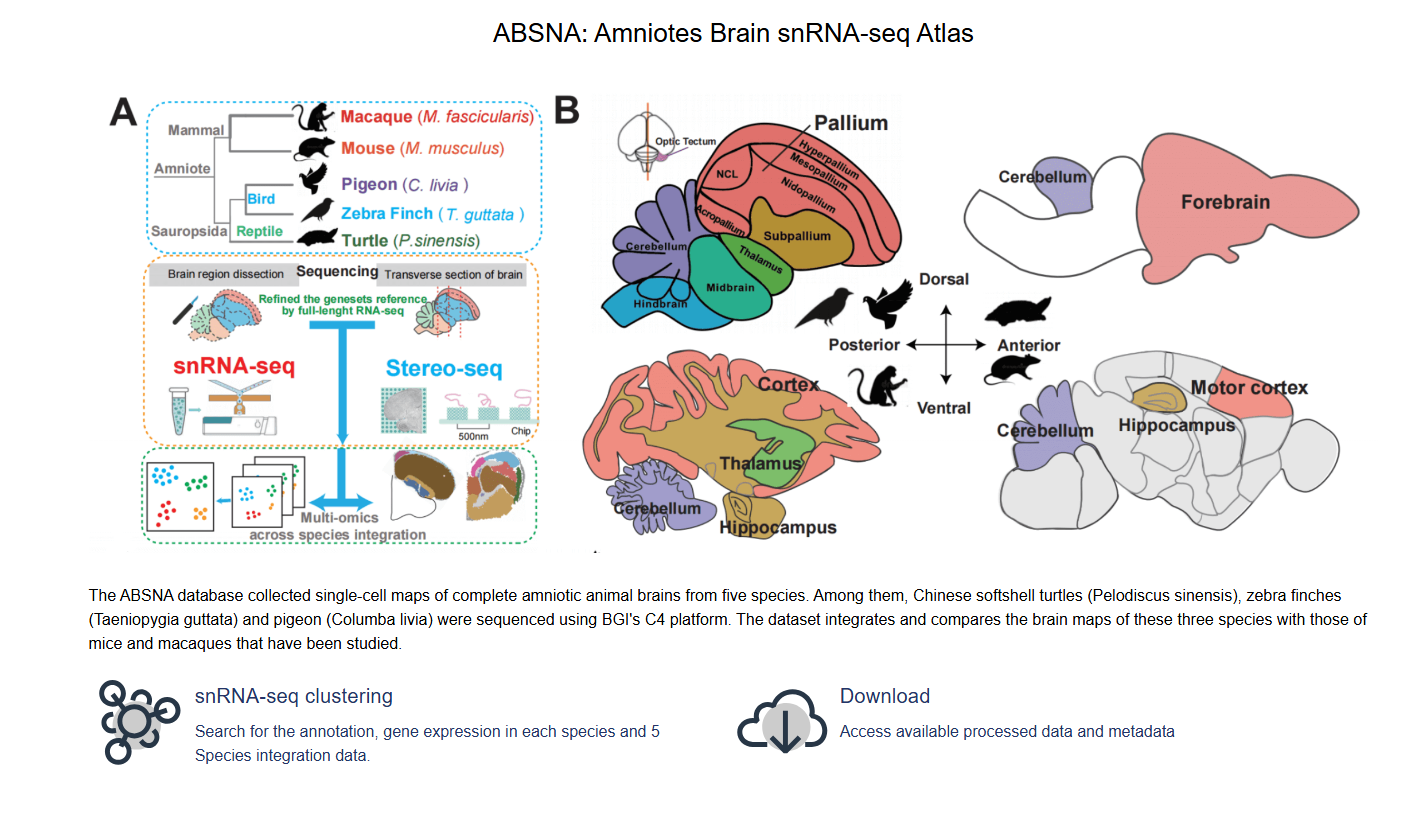
2025-07-07
Chen D, Zhuang Z, Huang M, Huang Y, Yan Y, Zhang Y, Lin Y, Jin X, Wang Y, Huang J, Xu W, Pan J, Wang H, Huang F, Liao K, Cheng M, Zhu Z, Bai Y, Niu Z, Zhang Z, Xiang Y, Wei X, Yang T, Zeng T, Dong Y, Lei Y, Sun Y, Wang J, Yang H, Sun Y, Cao G, Poo M, Liu L, Naumann RK, Xu C, Wang Z, Xu X, Liu S. Genomic evolution reshapes cell-type diversification in the amniote brain. Dev Cell. 2025 Jul 7;60(13):1900-1915.e5.
DOI:10.1016/j.devcel.2025.04.014
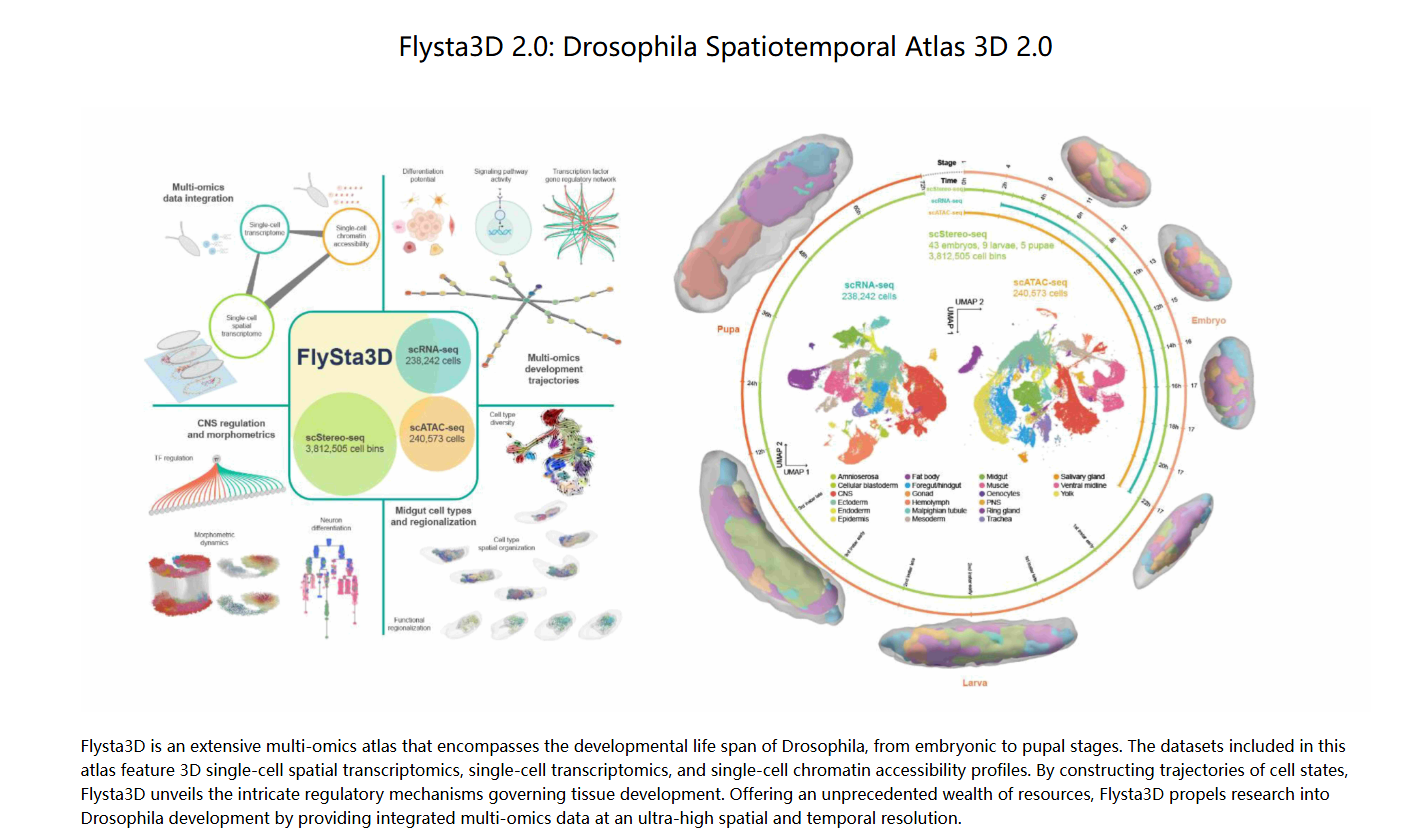
2025-08-21
Wang M, Hu Q, Tu Z, Kong L, Yu T, Jia Z, Wang Y, Yao J, Xiang R, Chen Z, Zhao Y, Zhou Y, Ye Q, Ouyang K, Wang X, Bai Y, Yang Z, Wang H, Wang Y, Jiang H, Yang T, Chen J, Huang Y, Yin N, Mo W, Liang W, Liu C, Lin X, Liu C, Gu Y, Chen W, Liu L, Xu X, Hu Y. A Drosophila single-cell 3D spatiotemporal multi-omics atlas unveils panoramic key regulators of cell-type differentiation. Cell. 2025 Aug 21;188(17):4734-4753.e31.
DOI::10.1016/j.cell.2025.05.04
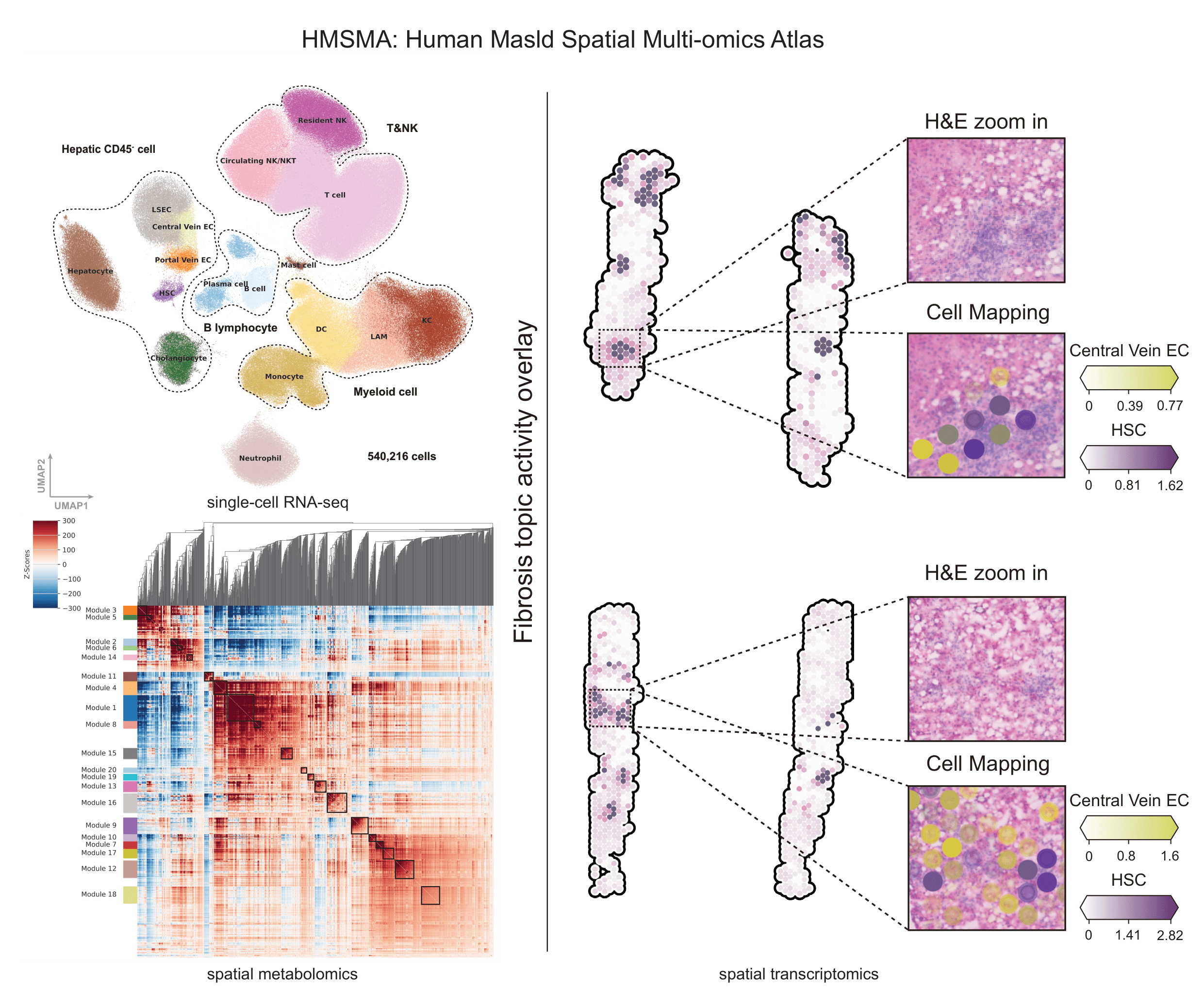
2025-11-24
Li, Z., Luo, G., Gan, C. et al. Spatially resolved multi-omics of human metabolic dysfunction-associated steatotic liver disease. Nat Genet 57, 3112–3125 (2025).
DOI:10.1038/s41588-025-02407-8
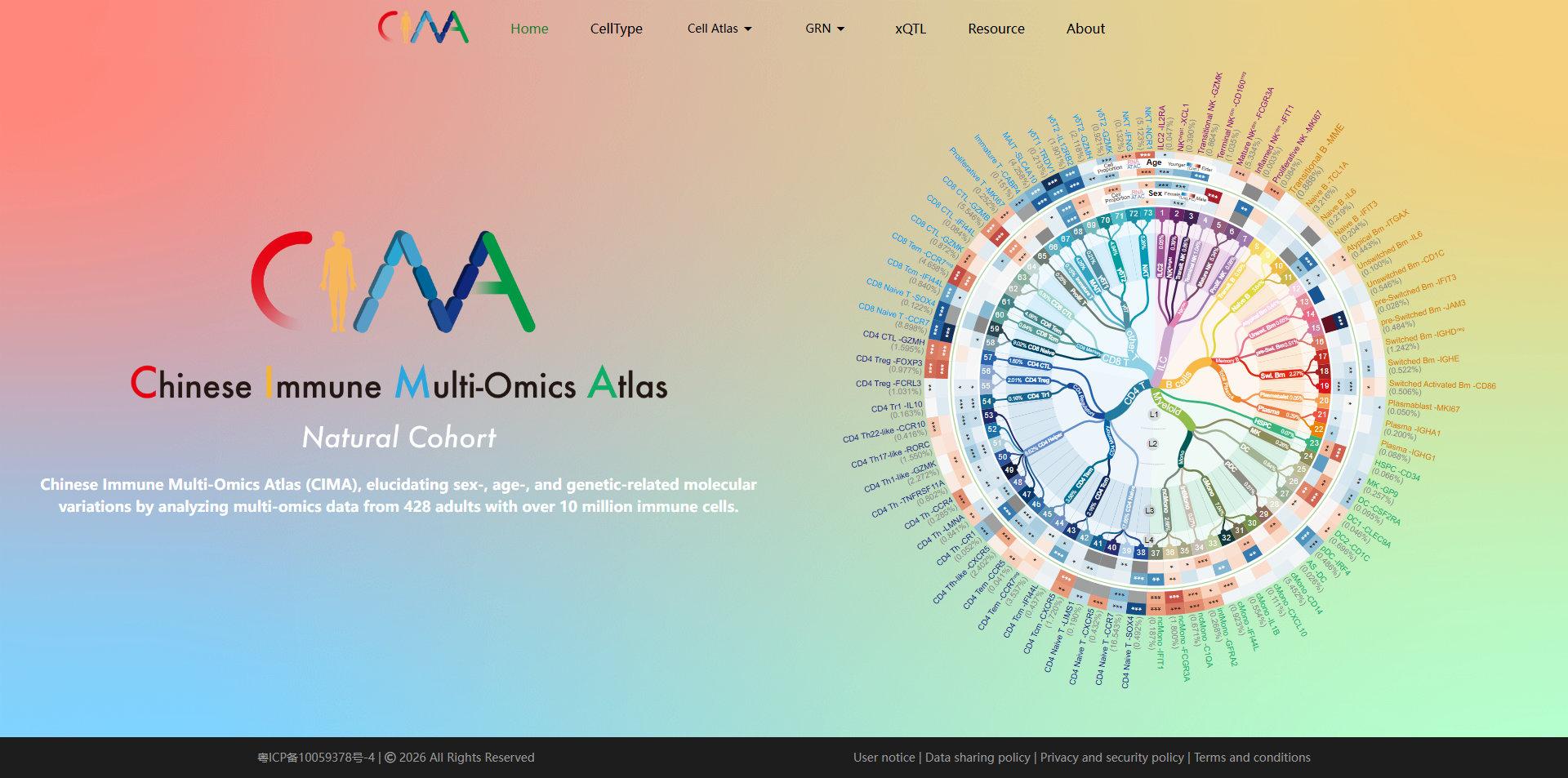
2026-01-08
Yin J, Zheng Y, Huang Z, Zhou W, Yuan Y, Cai P, Bai Y, Yang S, Gao Y, Duan S, Wang Y, Xu Z, Zhang W, Zhang X, Wei Y, Huang Y, Liu Y, Wang W, Yang T, Zhang Z, Chen X, Zhang X, Lv J, Li F, Zhang Y, Zeng G, Wang X, Ma W, Hou G, Hao S, Liu C, Lai Y, Liu P, Wang B, Li Y, Zhang W, Gao P, Xie J, Esteban MA, Gu Y, Liu X, Ji J, Qi T, Liu B, Wang H, Zhao Y, Yang X, Wang X, Chen R, Yang J, Yin Y, Wang J, Cao Y, Xu X, Liu L, Jin X, Liu C. Chinese Immune Multi-Omics Atlas. Science. 2026 Jan 8;391(6781):eadt3130.
DOI:10.1126/science.adt3130
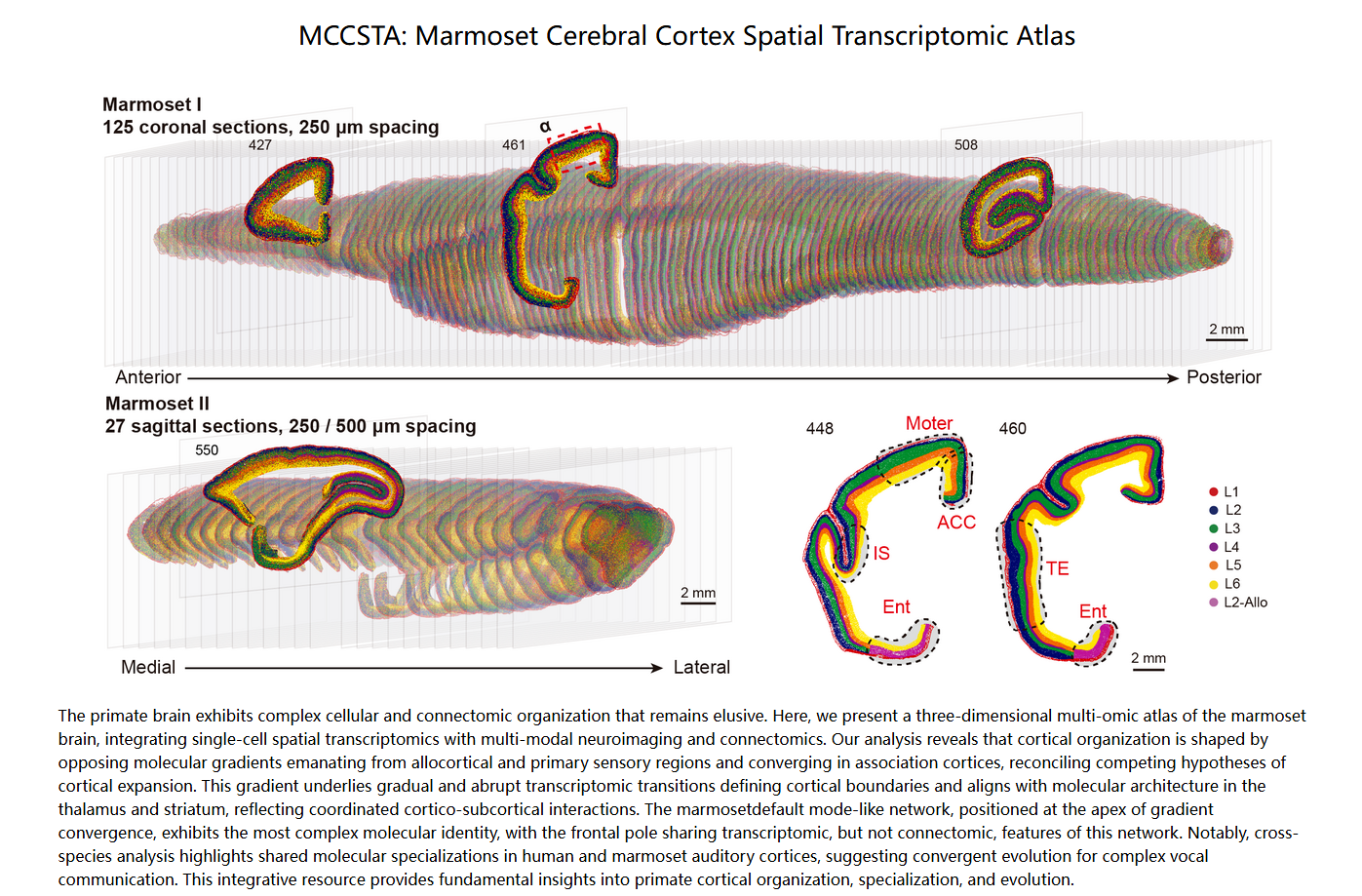
2026-04-16
Zhi Huang et al. ,An opposing molecular gradient axis underlies primate cortical organization.Science392,eaea2673(2026).
DOI:10.1126/science.aea2673
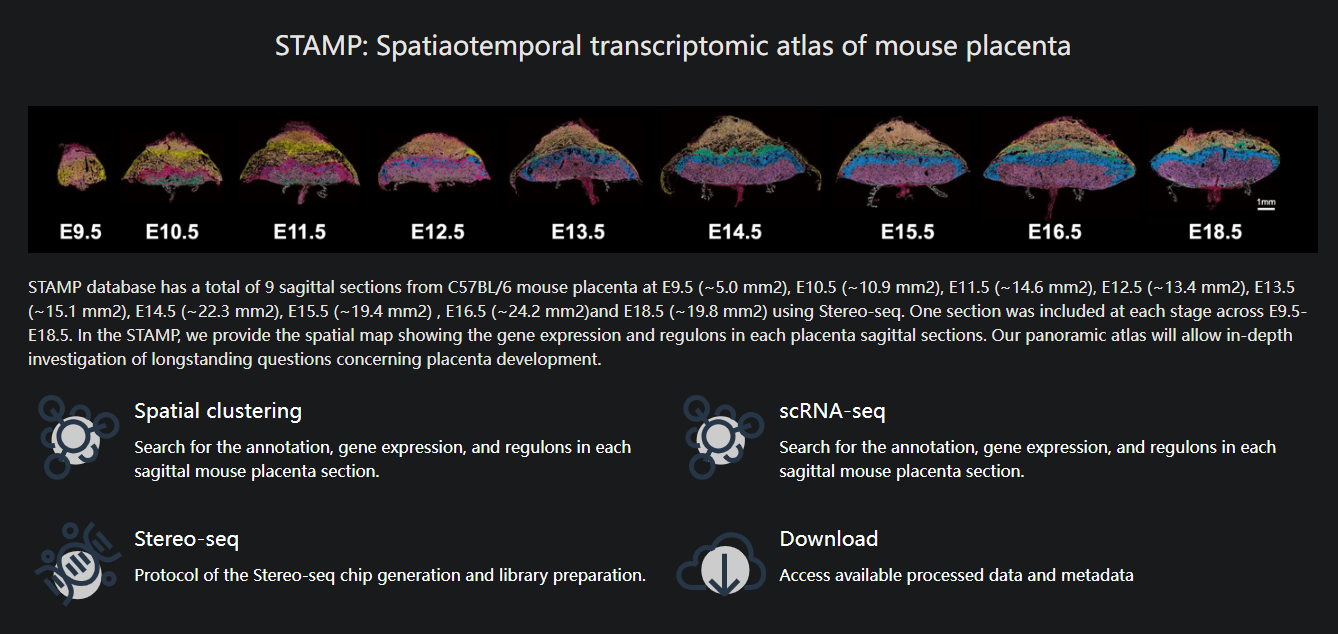
2026-05-01
Yuting FuXiaoqi ZengYifang LiuShikai JiaYujia JiangJia Ping TanYue YuanTianchang XiaYun MeiShan WenXiaojing LiuYue YouWeike PeiChengshuo YangSida ShaoJunhua ShenLiangshan MuXiaoxue MaMatthew Paul McCormackSaifeng ChengLuyi TianLongqi LiuXiaoyu WeiXiaodong Liu2026A spatiotemporal transcriptomic atlas of the mouse placenta reveals glycogen cell-mediated metabolic support essential for fetal viabilityeLife15:RP111257
DOI:10.7554/eLife.111257.1
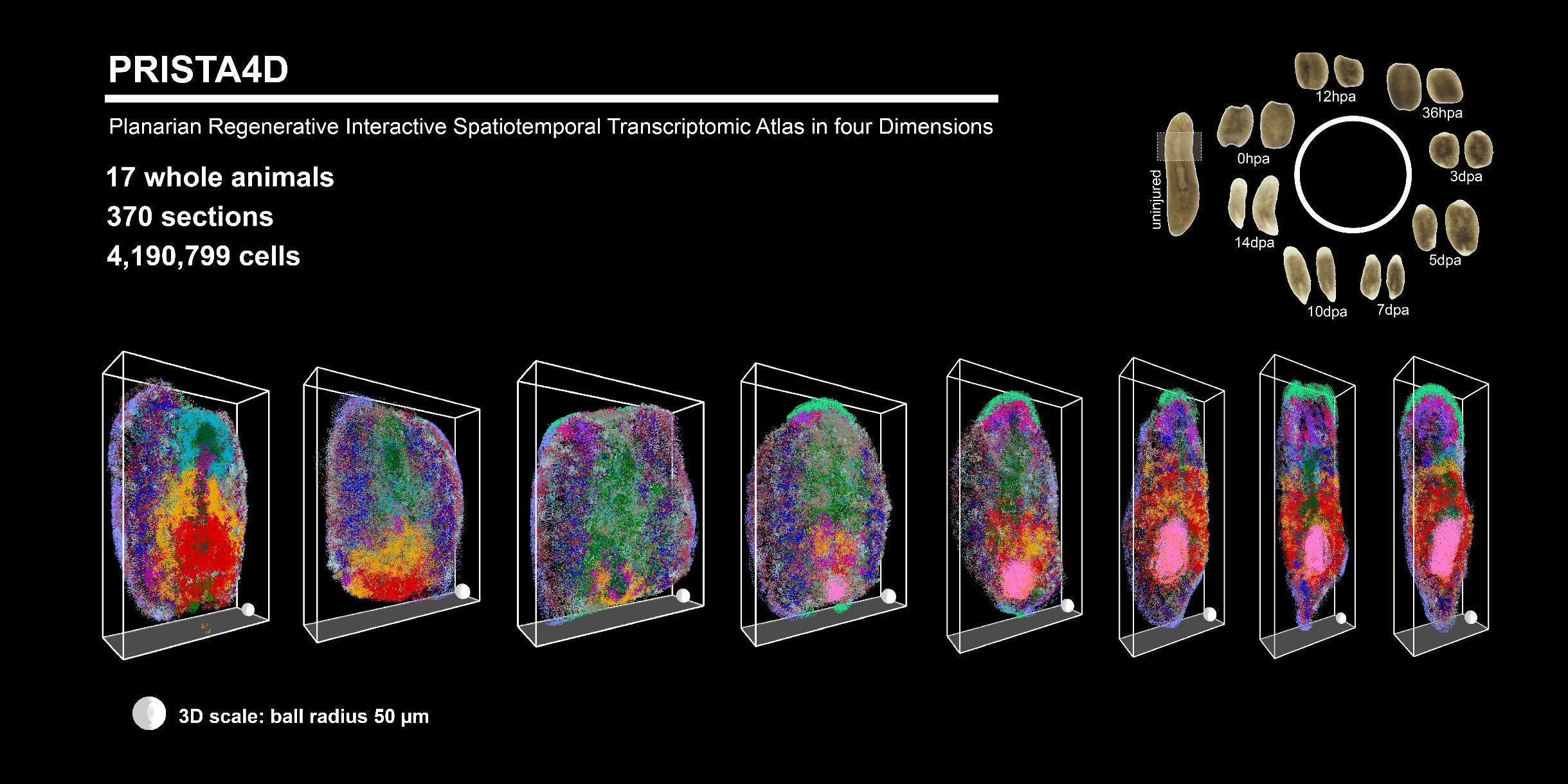
2026-05-22
Kai Han, Yuxiaofei Wang, Yao Li, Lidong Guo, Yue Chen, Xiawei Liu, Yaru Lin, Zhi Huang, Qun Liu, Wenjie Guo, Rui Zhang, Wandong Zhao, Langchao Liang, Xiaoyu Wei, Li Zhou, Xuebin Mao, Jiaqi Wang, Weijian Wu, Hongwei Pan, Tao Yang, He Zhang, Xiaoshan Su, Shanshan Liu, Wenwei Zhang, Longqi Liu, Søren Tvorup Christensen, Jifeng Fei, Xin Liu, Guangyi Fan, Hanbo Li, Ying Gu, Jian Wang, Huanming Yang, Gang Pei, Xun Xu, An Zeng, Mengyang Xu, 4D Single-Cell Spatial Transcriptomics Reveals Dynamic Morphogenetic Gradients and Regenerative Domains in Planarians, GigaScience, 2026;, giag064, https://doi.org/10.1093/gigascience/giag064
DOI:10.1093/gigascience/giag064
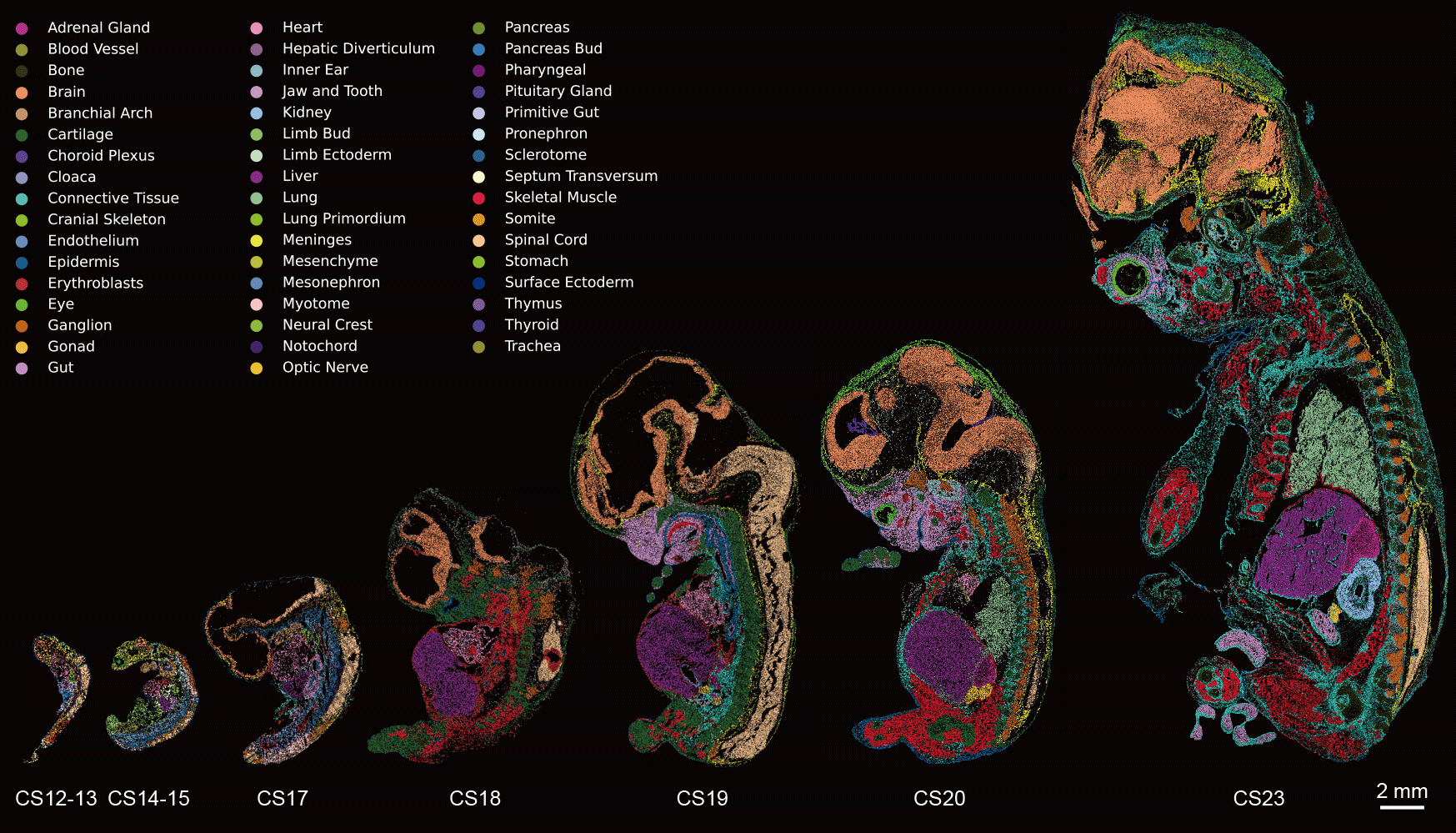
2026-05-27
Pan, J., Li, Y., Lin, Z. et al. Spatiotemporal transcriptome atlas of human embryos after gastrulation. Nature (2026).
DOI:10.1038/s41586-026-10545-0
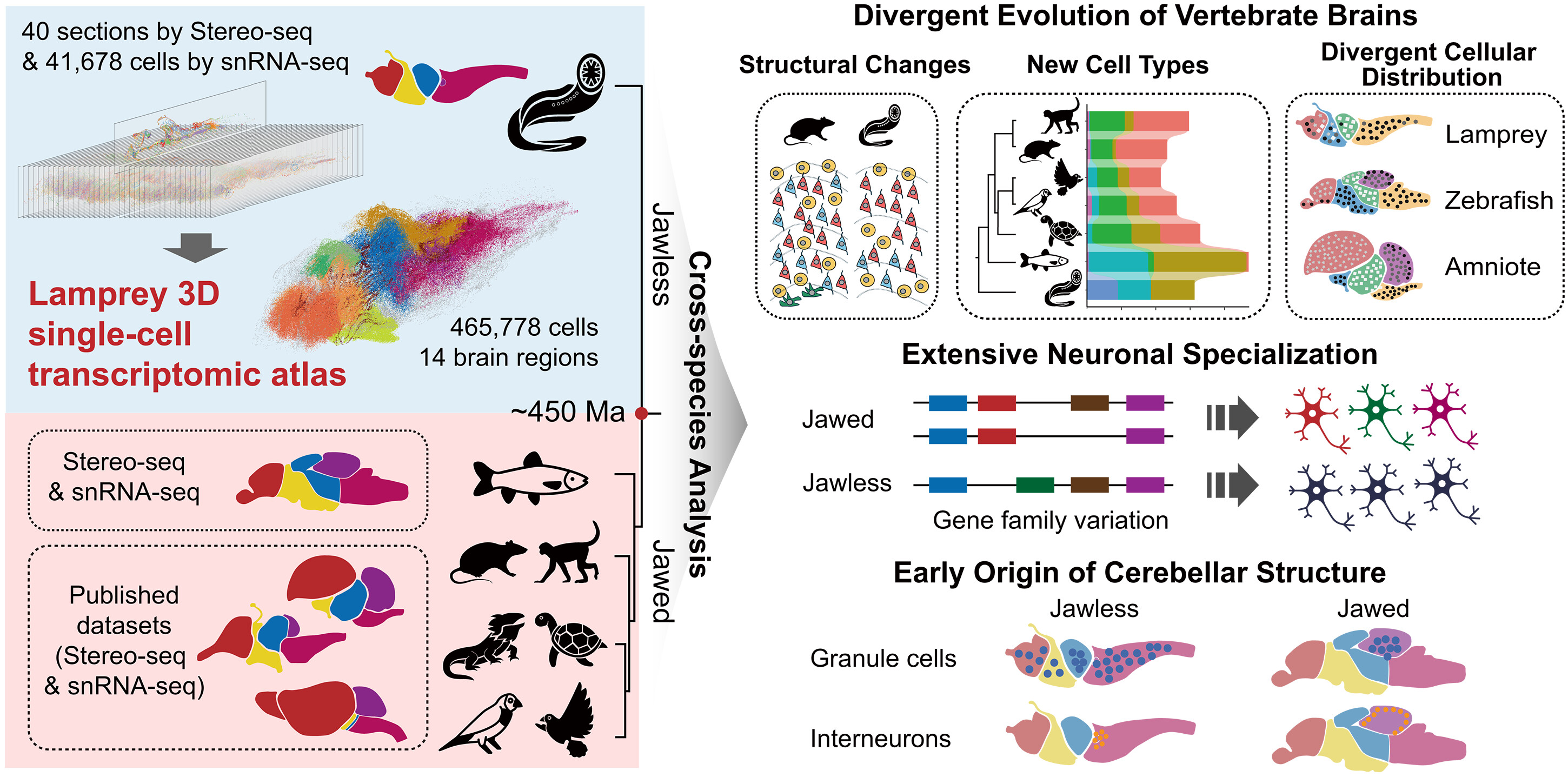
2026-06-18
Haixu Wu et al. ,Lamprey 3D single-cell transcriptomics reveals ancestral and specialized features of the vertebrate brain.Science392,eaea2535(2026).
DOI:10.1126/science.aea2535

2026-07-23
Shanshan Lian et al. ,Spatially resolved single-cell atlas reveals the macroevolutionary trajectory of animal hearts.Science0,eadw0855
DOI:10.1126/science.adw0855