chevron_leftchevron_right

We have only begun to scratch the surface in understanding mammalian development. An overwhelming caveat is the lack of topographical transcriptomic information to correlate signaling cues and cell-cell interactions within the hierarchy of cell fate decisions. Spatially resolved transcriptomic technologies are promising tools to fill this gap. However, the small field of view and imbalance between resolution and transcript capture of current methodologies precludes their systematic application to study relatively large and multilayered embryos. Here, we have combined DNA nanoball (DNB) patterned arrays and in situ RNA capture to create SpaTial Enhanced REsolution Omics-sequencing (Stereo-seq). This approach allows transcriptomic profiling of large histological sections with high resolution, sensitivity, and reproducibility. We have applied Stereo-seq to study the kinetics of transcriptional variation, the networks of transcription factor binding events and their relationship with morphogens across spatial domains of gene expression in a time course of mouse organogenesis. We have used this information to detect the emergence of tissue-specific cell identities such as early neuroblast populations in the late neural tube stage or the gradients of neuronal specification in the neocortex. Furthermore, we have mapped the expression of a panel of developmental disease-related loci to define the spatiotemporal windows of tissue vulnerability. Our panoramic atlas constitutes a essential resource to investigate longstanding questions concerning the molecular basis of normal and abnormal mammalian development.

2High-resolution spatiotemporal transcriptomic maps of developing Drosophila embryos and larvae
(ID: STDS0000060)
This database is intended to curate 3D spatial transcriptomes of all stages of Drosophila embryos and larvae generated by Stereo-seq. Drosophila melanogaster strain w1118 embryos were collected at two late stages (14-16 h and 16-18 h after egg laying, corresponding to stage 16~17 of embryogenesis) and all three stages were also collected. These samples were subject to cryosection to generate 7~10 μm thick slices. All slices of each sample were applied to Stereo-seq chips to capture their 2D spatial transcriptomes. All the 2D spatial transcriptomes of each sample were combined to recreate their 3D spatial transcriptomes. More samples of different stages will be added in the future. With these data, one could visualize and analyze spatial expression patterns of genes of interest, 3D reconstruct tissue-specific spatial transcriptomes by clustering and annotation, simulate tissue developmental trajectory across development, identify cell signalling pathways and gene regulatory networks, examine gene functions in their intact spatial context etc.
Mingyue Wang,Qinan Hu,Tianhang Lv,Yuhang Wang,Rong Xiang,Qing Lan,Zhencheng Tu,Yanrong Wei,Kai Han,Y

3ARTISTA: Axolotl Regenerative Telencehpalon Interpretation via Spatiotemporal Transcriptomic Atlas
(ID: STDS0000056)
Axolotl Regenerative Telencehpalon Interpretation via Spatiotemporal Transcriptomic Atlas (ARTISTA) is a spatially resolved transcriptomic data resource that provides visualization of gene expression across the regeneration and development stages of axolotl telencephalon at single cell resolution, aiming to provide a systematic dissection of the molecular events underlying neural regeneration in the axolotl brain, laying the foundation for further mechanistic studies. To comprehensively understand cellular dynamics occurred during axolotl brain regeneration and development, here, we carried out a series of spatial transcriptome analyses on serial sections along the rostral-caudal axis of 2 (3 sections), 5 (3 sections), 10 (3 sections), 15 (4 sections), 20 (3 sections), 30 and 60 days post injury (DPI) brain tissues after removal of a reproducible portion of dorsal pallium in left telencephalic hemisphere of 11 cm length axolotl. We also collected sections from developmental (stage 44, 54, and 57), juvenile, adult, and metamorphosed axolotl telencephalons. Based on this dataset, researchers can quickly explore the gene expression profiles of their interested cell types in spatial map across different regeneration and development stages of axolotl telencephalon.

Vertebrate embryogenesis is a remarkably dynamic process during which numerous cell types of different lineages generate, change, or disappear within a short period. A major challenge in understanding this process is the lack of topographical transcriptomic information that can help correlate microenvironmental cues within the hierarchy of cell fate decisions. Here, we employed Stereo-seq, a high-definition spatially resolved transcriptomic technology, to dissect the spatiotemporal dynamics of gene expression and regulatory networks in the developing zebrafish embryos. We profiled 91 embryo sections covering six critical time points during the first 24 hours of development, obtaining a total of 152,977 spots at a resolution of 10x10x15 µm3 (close to cellular size) with spatial coordinates. Meanwhile, we identified spatial modules and co-varying genes for specific tissue organizations. By performing the integrated analysis of the Stereo-seq and scRNA-seq data from each time point, we reconstructed the spatially resolved developmental trajectories of cell fate transitions and molecular changes during zebrafish embryogenesis. Our study constitutes a fundamental reference for further studies aiming to understand vertebrate development.

Recent discoveries about the molecular heterogeneity of the cerebellar cortex suggest the existence of functionally divergent subclasses of anatomically defined cell types. Using spatial transcriptome and single-nucleus RNA-seq analysis, we mapped 3D transcriptomic atlases of the whole cerebellum of mice, marmosets, and macaques at the single-cell resolution. Comparative analysis revealed specific cell types, cell localizations, and intra-cerebellum molecular heterogeneity across species. A comprehensive database generated from this study will expand the acknowledgment of the mammalian cerebellum.
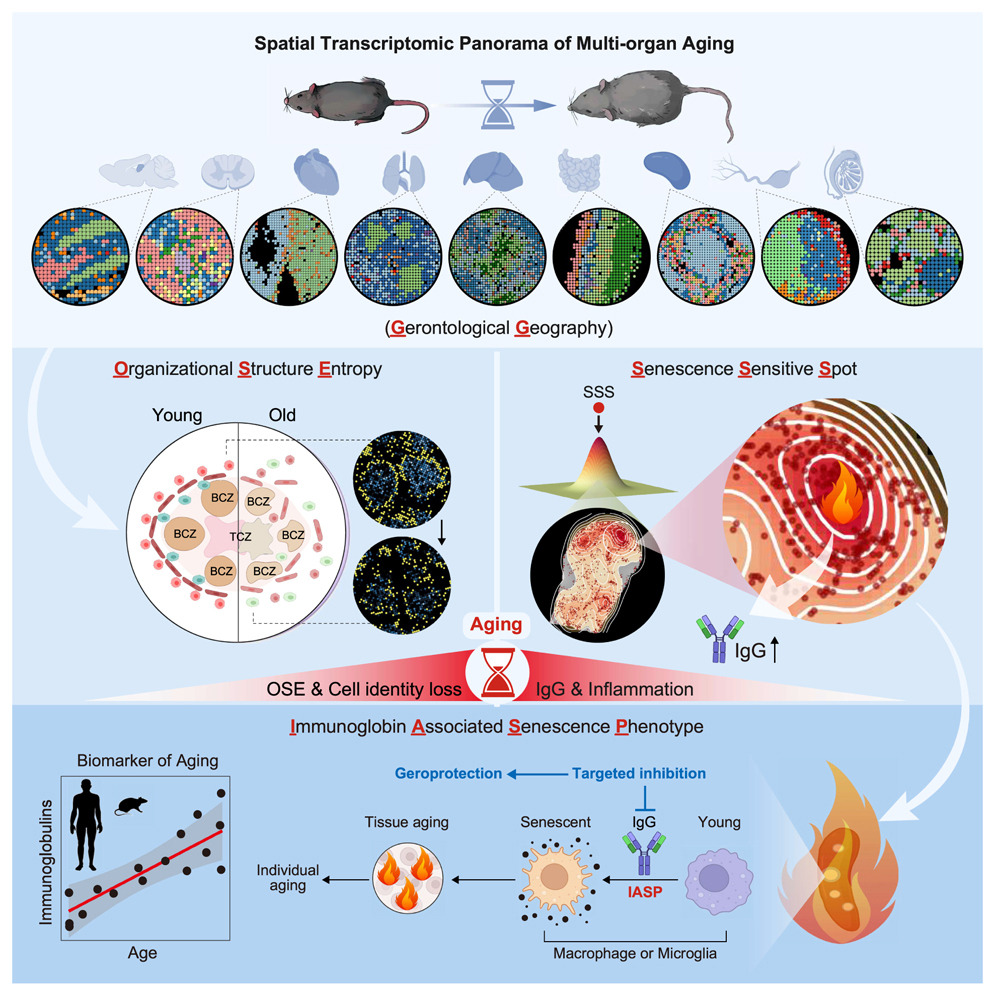
6Spatial transcriptomic landscape unveils immunoglobin-associated senescence as a hallmark of aging
(ID: STDS0000247)
To systematically characterize the loss of tissue integrity and organ dysfunction resulting from aging, we produced an in-depth spatial transcriptomic profile of nine tissues in male mice during aging. We showed that senescence-sensitive spots (SSSs) colocalized with elevated entropy in organizational structure and that the aggregation of immunoglobulin-expressing cells is a characteristic feature of the microenvironment surrounding SSSs. Immunoglobulin G (IgG) accumulated across the aged tissues in both male and female mice, and a similar phenomenon was observed in human tissues, suggesting the potential of the abnormal elevation of immunoglobulins as an evolutionarily conserved feature in aging. Furthermore, we observed that IgG could induce a pro-senescent state in macrophages and microglia, thereby exacerbating tissue aging, and that targeted reduction of IgG mitigated aging across various tissues in male mice. This study provides a high-resolution spatial depiction of aging and indicates the pivotal role of immunoglobulin-associated senescence during the aging process.
Yuzhe Sun

7Spatiotemporal analysis of human intestinal development at single-cell resolution
(ID: STDS0000002)
Development of the human intestine is not well understood. Here we link single-cell RNA sequencing and spatial transcriptomics to characterize intestinal morphogenesis through time. We identify 101 cell states including novel epithelial and mesenchymal progenitor populations and programs linked to key morphogenetic milestones. We describe principles of crypt-villus axis formation; neural, vascular, mesenchymal morphogenesis and immune population of the developing gut. We identify the differentiation hierarchies of developing fibroblast and myofibroblast subtypes and describe new functions for these including as vascular niche cells. We pinpoint the origins of Peyer’s patches and gut-associated lymphoid tissue (GALT) and describe location specific immune programs. We use our resource to present an unbiased analysis of morphogen gradients that direct sequential waves of cellular differentiation and define cells and locations linked to rare developmental intestinal disorders.
FawknerCorbett D,Antanaviciute A,Parikh K,Jagielowicz M,Sousa Gerós A,Gupta T,Ashley N,Koohy H,Simmo
![Spatial transcriptomics combined with single-cell RNA-sequencing unravels the complex inflammatory cell network in atopic dermatitis [ST]](/stomics/media/dataset/dataset/image/STDS0000212.png)
8Spatial transcriptomics combined with single-cell RNA-sequencing unravels the complex inflammatory cell network in atopic dermatitis [ST]
(ID: STDS0000212)
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease with complex pathogenesis. Using spatial and single-cell transcriptomics of whole skin biopsy and suction blister material, we investigated the cellular and molecular features of the leukocyte-infiltrated area in AD. We identified unique clusters of fibroblasts, dendritic cells, macrophages, and T cells in the lesional AD skin and molecular interactions between these cells. The leukocyte-infiltrated areas in lesional AD skin showed upregulation of COL6A5, COL4A1, TNC, IL32, CCL19 in COL18A1-expressing fibroblasts. Additionally, M2 macrophages expressed CCL13 and CCL18 in the same localization. Ligand–receptor interaction analysis of the spatial transcriptome identified a neighboring infiltration and interaction between activated COL18A1-expressing fibroblasts, activated CCL13- and CCL18-expressing M2 macrophages, CCR7- and LAMP3-expressing DCs, and T cells. As observed in skin lesions, serum levels of TNC and CCL18 were significantly elevated in AD and correlated with clinical disease severity.
Mitamura, Yasutaka,Reiger, Matthias,Kim, Juno,Xiao, Yi,Zhakparov, Damir,Tan, Ge,Rinaldi, Arturo O,Ba
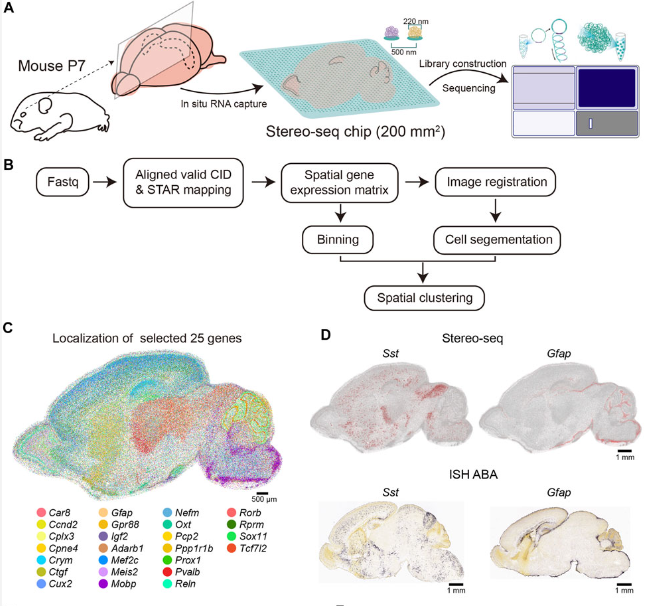
9A cellular resolution spatial transcriptomic landscape of the postnatal mouse brain
(ID: STDS0000139)
Here we apply Stereo-seq to generate a spatially-resolved transcriptomic description of the postnatal day 7 (P7) murine whole brain sagittal section near the middle line. Our study comprehensively dissected the anatomical regions, gene expression and gene regulatory network patterns and cell type localization at whole brain scale.
Mengnan cheng; Liang Wu; Chuanyu Liu; Longqi Liu

10The Single-cell Stereo-seq reveals region-specific cell subtypes and transcriptome profiling in Arabidopsis leaves
(ID: STDS0000104)
We present the first in situ single-cell transcriptome profiling in plant, scStereo-seq (single-cell SpaTial Enhanced REsolution Omics-sequencing), which enabled the bona fide single-cell spatial transcriptome of Arabidopsis leaves.
Keke Xia,Hai-Xi Sun,Jie Li,Jiming Li,Yu Zhao,Lichuan Chen,Chao Qin,Ruiying Chen,Guangyu Liu,Zhiyong