Introduction
Introduction
BLAST stands for Basic Local Alignment Search Tool. The BLAST service of CNGB is developed with NCBI BLAST+ 2.8.1 standalone version, downloaded from NCBI FTP server, providing sequences searching on public data of CNGB applications, BGI projects and external data sources.
The word, BLAST, in the name "the BLAST service of CNGB", is standing for kinds of sequence searching. More types of sequence searching will be integrated in the future.
Usage
Usage
1. System requirement
It is recommended to use the latest version of Firefox or Chrome browser to access the BLAST service of CNGB.
2. Homepage of BLAST
The homepage (https://db.cngb.org/blast/) of BLAST contains several search services, including BLASTN, BLASTP, BLASTX, TBLASTN, TBLAST.
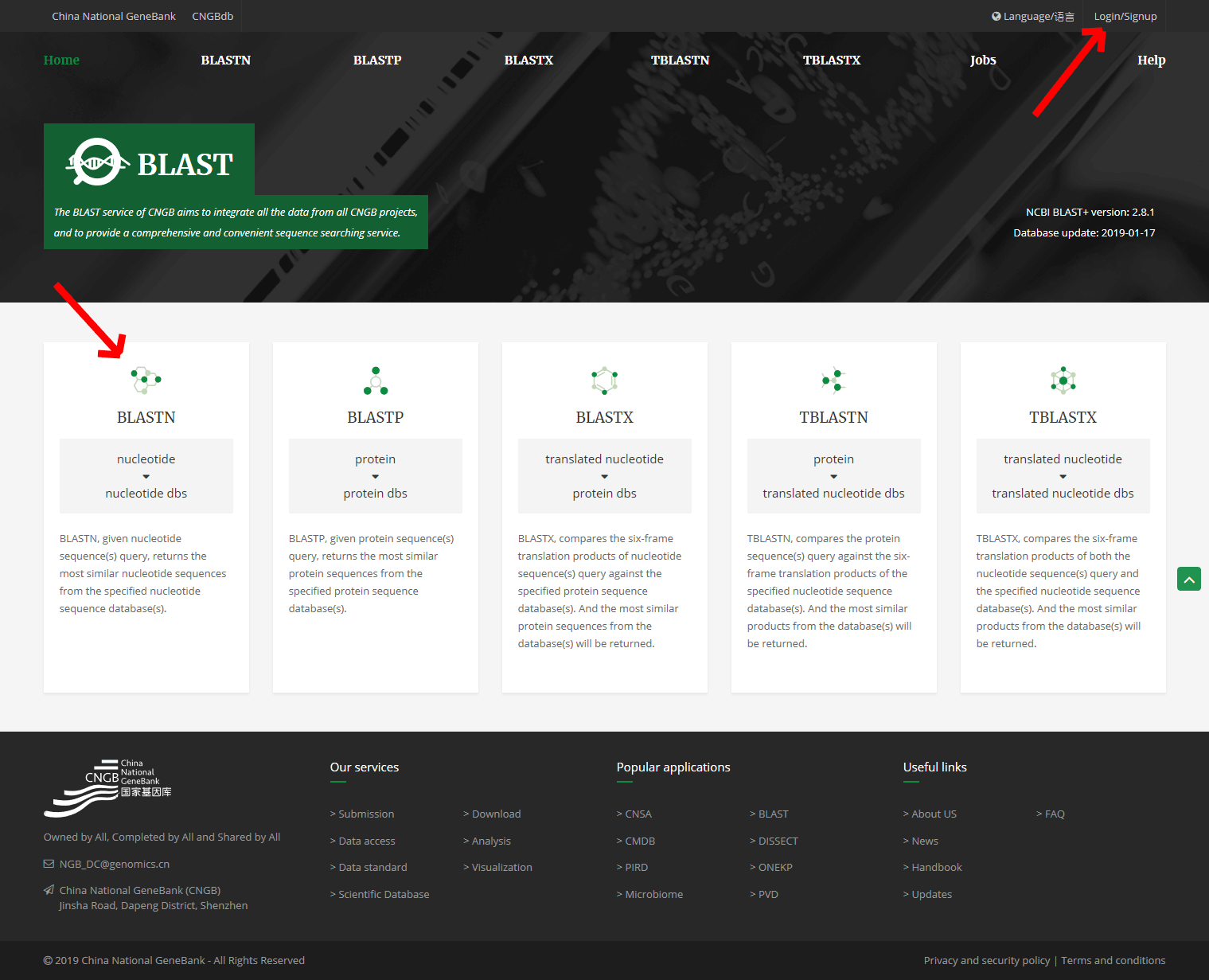
3. Registration and login
All CNGB database Service use a unified user registration and login platform. The registered account applies to the BLAST search. Click the tab of "LOGIN/SIGNUP" on the right side of the homepage to enter the registration and login page.
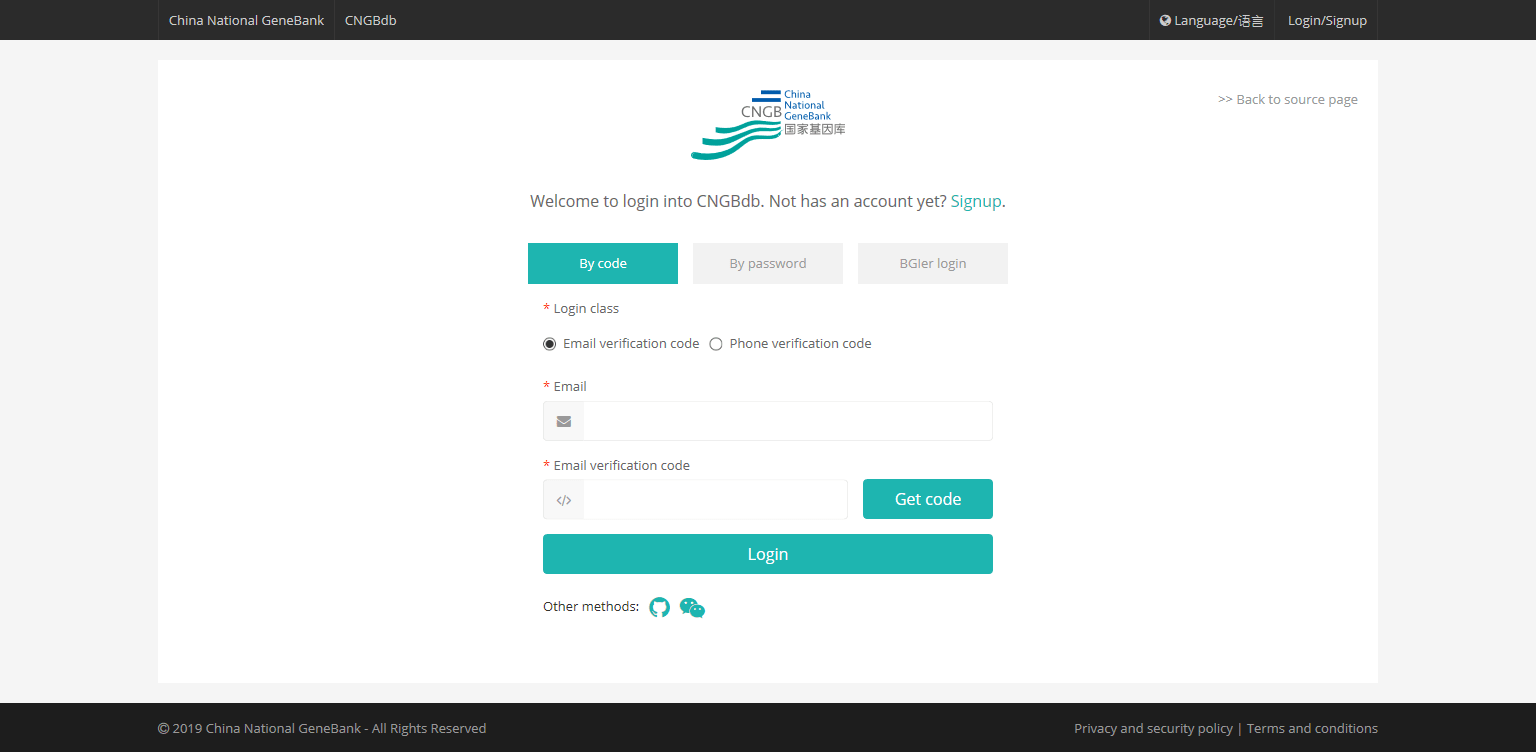
All jobs and its' results which are completed will be saved for 3 days in the job list.
If you use the BLAST search service directly, you only can see the currently submitted job in the job list. Please record the job code or job title by yourself, and you could use them to search in the 「jobs」 page in 3 days.
4. Submit data to BLAST search
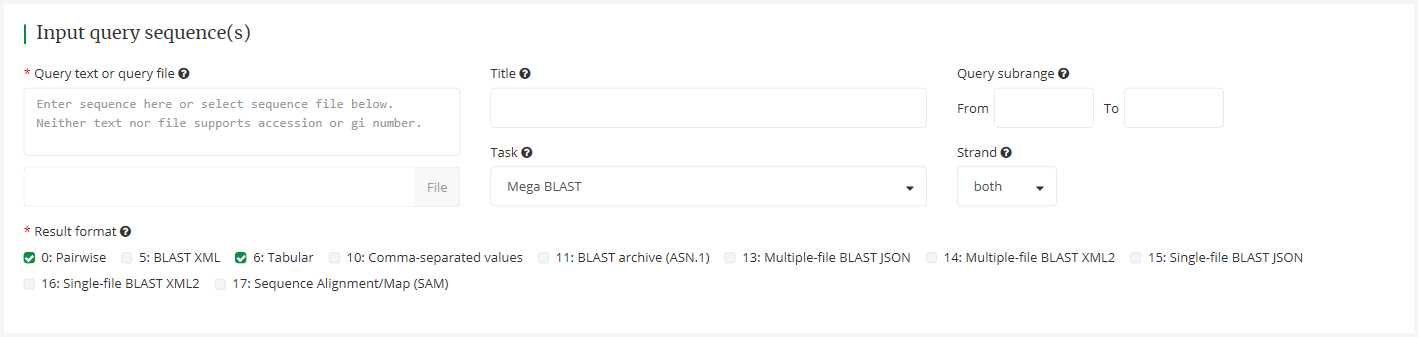
4.1 Input the sequences and other related information
Please input sequence or upload the fasta sequence file (required), and fill other related information about the sequences (Optional) . Then please choose at least one of results file formats (required, the first format is selected by default).
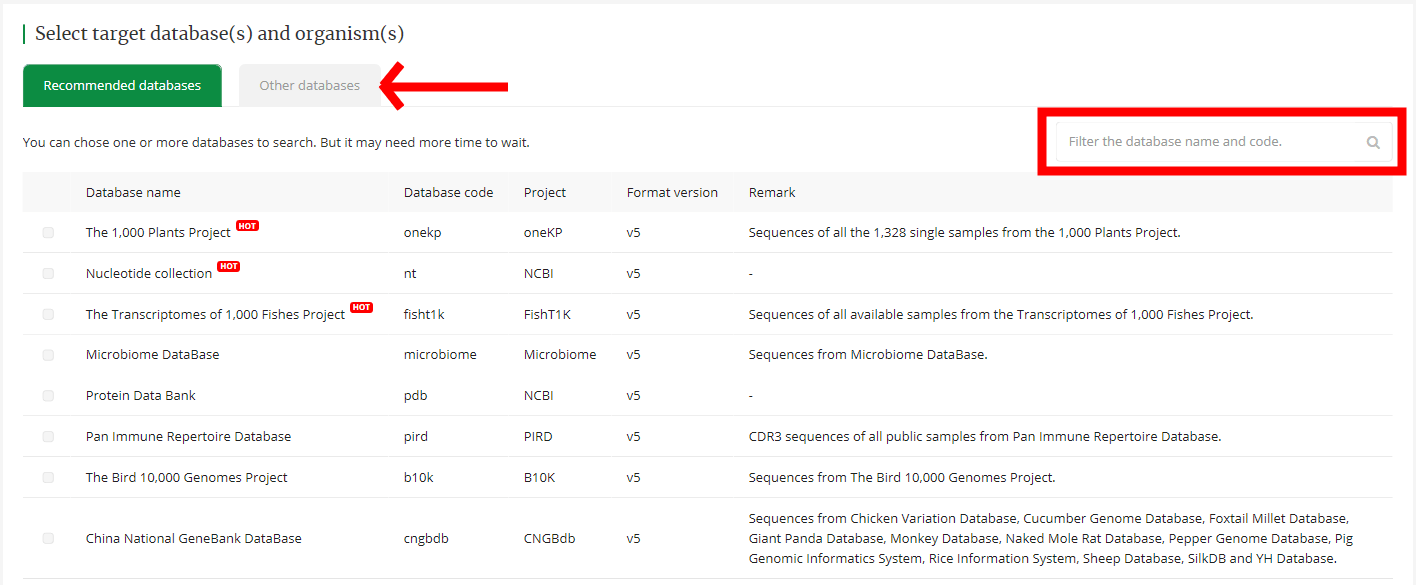
4.2 Select the database for search
We provide two types of database (recommended database and other database), and there is a search box on the right side of the database list to seek your target database. Then You could select one or more database for the BLAST search.
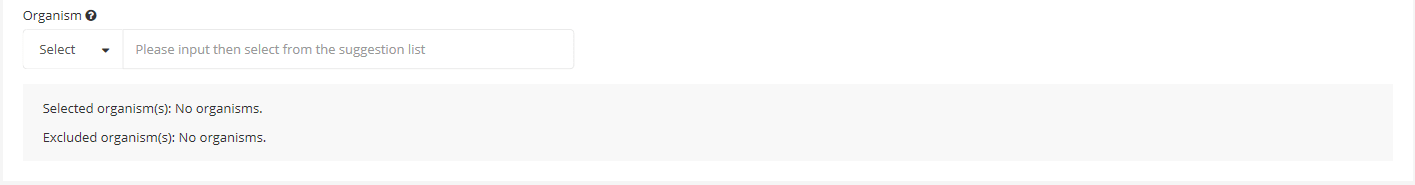
4.3 Organism setting
You can 「Select」 or 「Exclude」 the specific organisms to BLAST search.
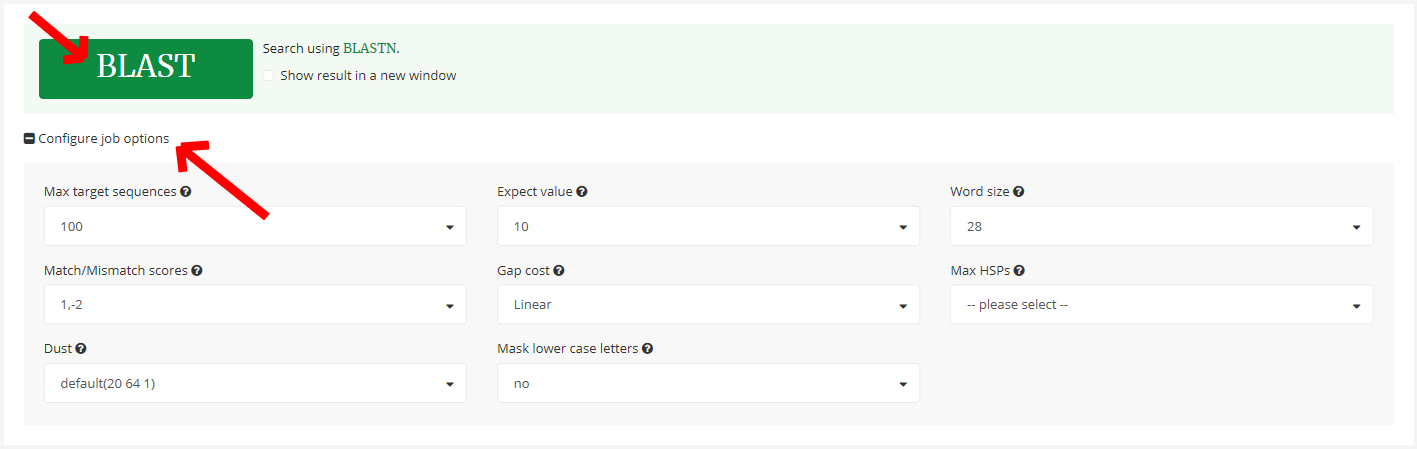
4.4 Submit BLAST search and more job options
Click 「BLAST」 button to submit the BLAST search job.
If you have some special requirements with the search results, you can click 「Configure job options」 to tune options of the search job.
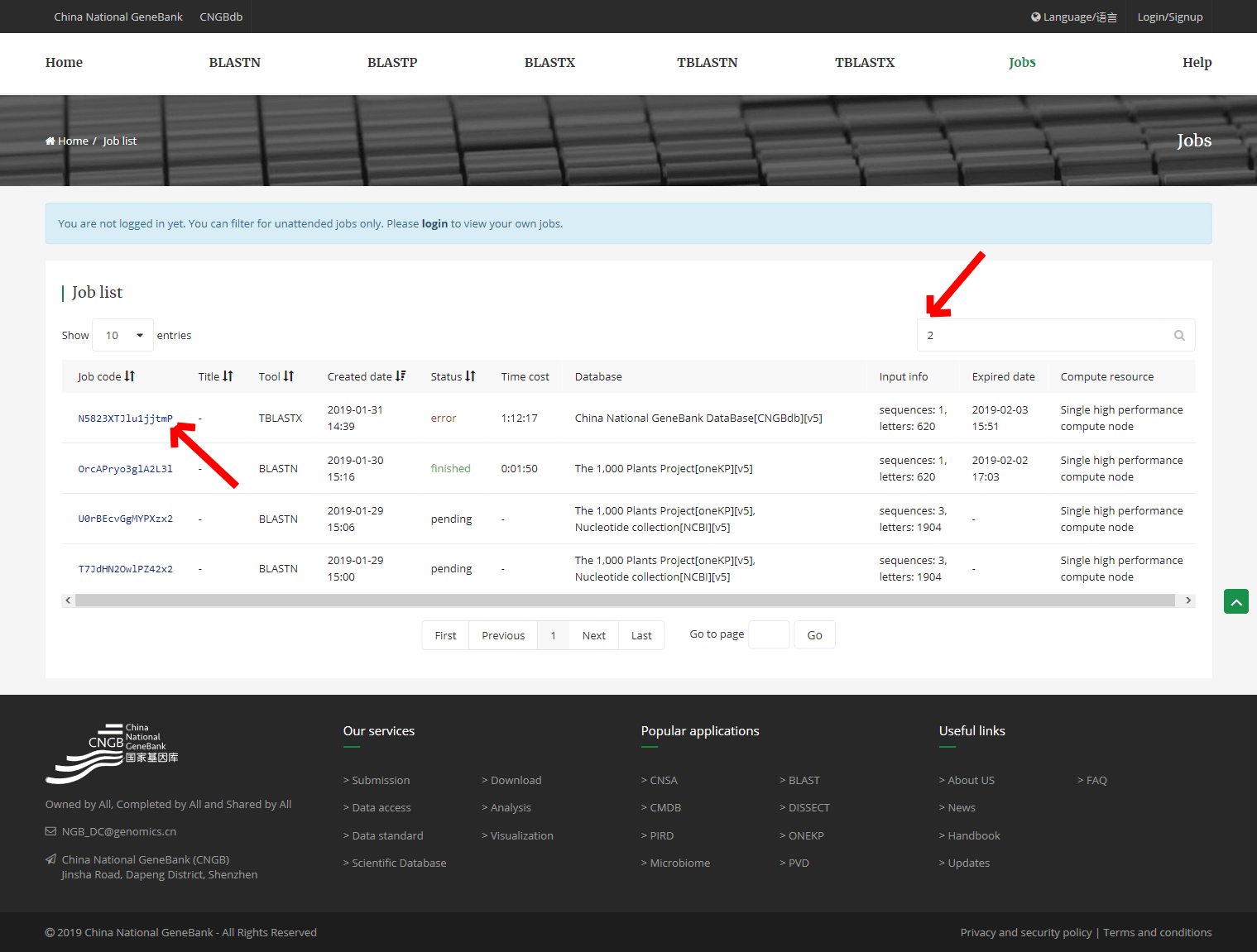
5. Job list
When you submit the BLAST search job, the page will automatically redirect to the 「job」 page. And you can click the 「Job code」 into the results detail page for viewing and downloading the result.
If you have been logged in, your all job and its results which is completed will be saved for 3 days on the 「Job」 page’s list.
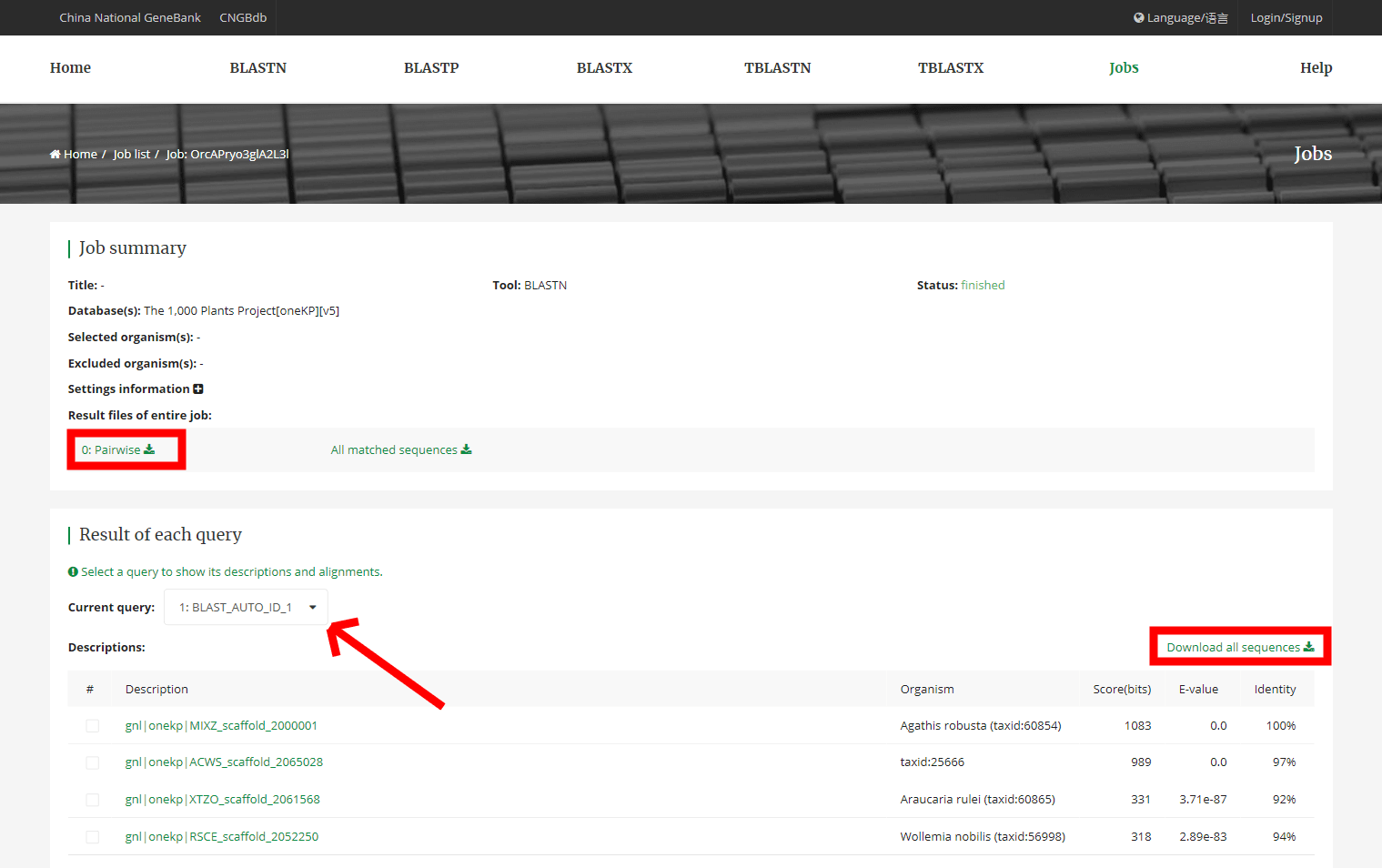
6. BLAST search results
The BLAST search results can be viewed on the detail page. Also You can download the results files which format you have been selected and the sequence file.
Documentation
Documentation
Query input
The query sequence(s) to be used for a BLAST search should be pasted in the "Query text" text area. Or saved as a file and provided through the "Query file". If both "Query text" and "Query file" are filled, the content from these two fields will be simply combined together as query input.
BLAST accepts a number of different types of input and automatically determines the format or the input. Accepted input types are FASTA or bare sequence.
FASTA
A sequence in FASTA format begins with a single-line description, followed by lines of sequence data. The description line (defline) is distinguished from the sequence data by a greater-than (">") symbol at the beginning. It is recommended that all lines of text be shorter than 80 characters in length. An example sequence in FASTA format is:
>P01013 GENE X PROTEIN (OVALBUMIN-RELATED)
QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE
KMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTS
VLMALGMTDLFIPSANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRADHP
FLFLIKHNPTNTIVYFGRYWSPBlank lines are not allowed in the middle of FASTA input.
Sequences are expected to be represented in the standard IUB/IUPAC amino acid and nucleic acid codes, with these exceptions: lower-case letters are accepted and are mapped into upper-case; a single hyphen or dash can be used to represent a gap of indeterminate length; and in amino acid sequences, U and * are acceptable letters (see below). Before submitting a request, any numerical digits in the query sequence should either be removed or replaced by appropriate letter codes (e.g., N for unknown nucleic acid residue or X for unknown amino acid residue). The nucleic acid codes supported are:
A adenosine C cytidine G guanine
T thymidine N A/G/C/T (any) U uridine
K G/T (keto) S G/C (strong) Y T/C (pyrimidine)
M A/C (amino) W A/T (weak) R G/A (purine)
B G/T/C D G/A/T H A/C/T
V G/C/A - gap of indeterminate lengthFor those programs that use amino acid query sequences (BLASTP and TBLASTN), the accepted amino acid codes are:
A alanine P proline
B aspartate/asparagine Q glutamine
C cystine R arginine
D aspartate S serine
E glutamate T threonine
F phenylalanine U selenocysteine
G glycine V valine
H histidine W tryptophan
I isoleucine Y tyrosine
K lysine Z glutamate/glutamine
L leucine X any
M methionine * translation stop
N asparagine - gap of indeterminate lengthBare sequence
This may be just lines of sequence data, without the FASTA definition line, e.g.:
QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE
KMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTS
VLMALGMTDLFIPSANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRADHP
FLFLIKHNPTNTIVYFGRYWSPIt can also be sequence interspersed with numbers and/or spaces, such as the sequence portion of a GenBank/GenPept flatfile report:
1 qikdllvsss tdldttlvlv naiyfkgmwk tafnaedtre mpfhvtkqes kpvqmmcmnn
61 sfnvatlpae kmkilelpfa sgdlsmlvll pdevsdleri ektinfeklt ewtnpntmek
121 rrvkvylpqm kieekynlts vlmalgmtdl fipsanltgi ssaeslkisq avhgafmels
181 edgiemagst gviedikhsp eseqfradhp flflikhnpt ntivyfgryw spBlank lines are not allowed in the middle of bare sequence input.
Database and organism
Detail to follow.
Query subrange
A segment of the query sequences can be used in BLAST searching. You can enter the range in the "Query subrange" box to specify the position of this segment. For example to limit matches to the region from 24 (the start) to 200 (the stop) of a query sequence, you would enter "24-200" in the field. If one of the limits you enter is out of range, the intersection of the [start,stop] and [1,length] intervals will be searched, where length is the length of the whole query sequence.
Genetic code
Genetic code to be used in blastx and tblastx translation of the query. Please refer to this NCBI page.
Word size
BLAST is a heuristic that works by finding word-matches between the query and database sequences. One may think of this process as finding "hot-spots" that BLAST can then use to initiate extensions that might eventually lead to full-blown alignments. For nucleotide-nucleotide searches (i.e., "blastn") an exact match of the entire word is required before an extension is initiated, so that one normally regulates the sensitivity and speed of the search by increasing or decreasing the word-size. For other BLAST searches non-exact word matches are taken into account based upon the similarity between words. The amount of similarity can be varied.
Expect value
This setting specifies the statistical significance threshold for reporting matches against database sequences. The default value (default is 10) means that 10 such matches are expected to be found merely by chance, according to the stochastic model of Karlin and Altschul (1990). If the statistical significance ascribed to a match is greater than the expect value, the match will not be reported. Lower expect values are more stringent, leading to fewer chance matches being reported.
Matrix
A key element in evaluating the quality of a pairwise sequence alignment is the "substitution matrix", which assigns a score for aligning any possible pair of residues. The matrix used in a BLAST search can be changed depending on the type of sequences you are searching with.
Match/Mismatch scores
Many nucleotide searches use a simple scoring system that consists of a "reward" for a match and a "penalty" for a mismatch. The (absolute) reward/penalty ratio should be increased as one looks at more divergent sequences. A ratio of 0.33 (1/-3) is appropriate for sequences that are about 99% conserved; a ratio of 0.5 (1/-2) is best for sequences that are 95% conserved; a ratio of about one (1/-1) is best for sequences that are 75% conserved (States et al., 1991).
Composition based stats
Amino acid substitution matrices may be adjusted in various ways to compensate for the amino acid compositions of the sequences being compared. The simplest adjustment is to scale all substitution scores by an analytically determined constant, while leaving the gap scores fixed; this procedure is called "composition-based statistics" (Schaffer et al., 2001). The resulting scaled scores yield more accurate E-values than standard, unscaled scores. A more sophisticated approach adjusts each score in a standard substitution matrix separately to compensate for the compositions of the two sequences being compared (Yu et al., 2003; Yu and Altschul, 2005; Altschul et al., 2005). Such "compositional score matrix adjustment" may be invoked only under certain specific conditions for which it has been empirically determined to be beneficial (Altschul et al., 2005); under all other conditions, composition-based statistics are used. Alternatively, compositional adjustment may be invoked universally.
Gap cost
The pull down menu shows the Gap Costs for the chosen Matrix. There can only be a limited number of options for these parameters. Increasing the Gap Costs will result in alignments which decrease the number of Gaps introduced.
DUST/SEG
This function mask off segments of the query sequence that have low compositional complexity, as determined by the SEG program of Wootton and Federhen (Computers and Chemistry, 1993) or, for BLASTN, by the DUST program of Tatusov and Lipman. Filtering can eliminate statistically significant but biologically uninteresting reports from the blast output (e.g., hits against common acidic-, basic- or proline-rich regions), leaving the more biologically interesting regions of the query sequence available for specific matching against database sequences.
Filtering is only applied to the query sequence (or its translation products), not to database sequences. Default filtering is DUST for BLASTN, SEG for other programs.
Mask lower case letters
With this option selected you can cut and paste a FASTA sequence in upper case characters and denote areas you would like filtered with lower case. This allows you to customize what is filtered from the sequence during the comparison to the BLAST databases.
Reference
Reference
- CNL_PMID20003500: Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. Published 2009 Dec 15. doi:10.1186/1471-2105-10-421
- CNL_PMID20003500: Altschul S, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. Published 1990 October 5. doi:10.1016/S0022-2836(05)80360-2
- 0755-33945586
- CNGBdb@cngb.org
- China National GeneBank (CNGB)
Jinsha Road, Dapeng District, Shenzhen
Resource
CNGBdb Wechat
