chevron_leftchevron_right

1Spatiotemporal analysis of human intestinal development at single-cell resolution
(ID: STDS0000002)
Development of the human intestine is not well understood. Here we link single-cell RNA sequencing and spatial transcriptomics to characterize intestinal morphogenesis through time. We identify 101 cell states including novel epithelial and mesenchymal progenitor populations and programs linked to key morphogenetic milestones. We describe principles of crypt-villus axis formation; neural, vascular, mesenchymal morphogenesis and immune population of the developing gut. We identify the differentiation hierarchies of developing fibroblast and myofibroblast subtypes and describe new functions for these including as vascular niche cells. We pinpoint the origins of Peyer’s patches and gut-associated lymphoid tissue (GALT) and describe location specific immune programs. We use our resource to present an unbiased analysis of morphogen gradients that direct sequential waves of cellular differentiation and define cells and locations linked to rare developmental intestinal disorders.
FawknerCorbett D,Antanaviciute A,Parikh K,Jagielowicz M,Sousa Gerós A,Gupta T,Ashley N,Koohy H,Simmo
![Spatial transcriptomics combined with single-cell RNA-sequencing unravels the complex inflammatory cell network in atopic dermatitis [ST]](/stomics/media/dataset/dataset/image/STDS0000212.png)
2Spatial transcriptomics combined with single-cell RNA-sequencing unravels the complex inflammatory cell network in atopic dermatitis [ST]
(ID: STDS0000212)
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease with complex pathogenesis. Using spatial and single-cell transcriptomics of whole skin biopsy and suction blister material, we investigated the cellular and molecular features of the leukocyte-infiltrated area in AD. We identified unique clusters of fibroblasts, dendritic cells, macrophages, and T cells in the lesional AD skin and molecular interactions between these cells. The leukocyte-infiltrated areas in lesional AD skin showed upregulation of COL6A5, COL4A1, TNC, IL32, CCL19 in COL18A1-expressing fibroblasts. Additionally, M2 macrophages expressed CCL13 and CCL18 in the same localization. Ligand–receptor interaction analysis of the spatial transcriptome identified a neighboring infiltration and interaction between activated COL18A1-expressing fibroblasts, activated CCL13- and CCL18-expressing M2 macrophages, CCR7- and LAMP3-expressing DCs, and T cells. As observed in skin lesions, serum levels of TNC and CCL18 were significantly elevated in AD and correlated with clinical disease severity.
Mitamura, Yasutaka,Reiger, Matthias,Kim, Juno,Xiao, Yi,Zhakparov, Damir,Tan, Ge,Rinaldi, Arturo O,Ba
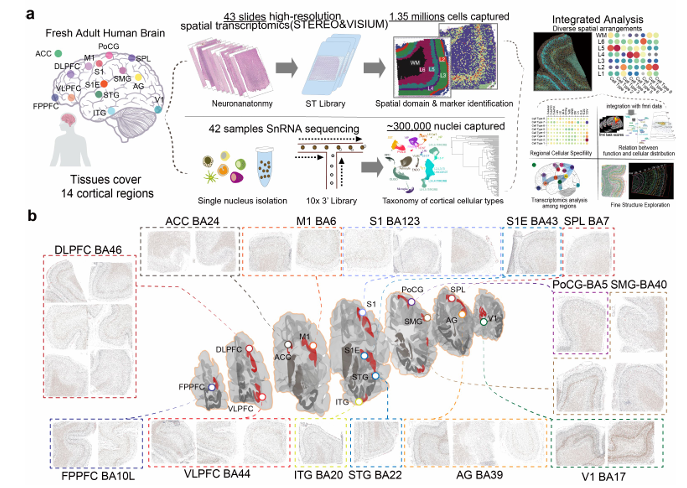
The higher-order cognitive functions of the human cortex rely on complex cellular structures, with diversity and spatial organization critical for specific functions. However, the molecular features and precise spatial organization of neural cells within the human cortex remain incompletely understood. Subcellular-level spatial transcriptomic sequencing and snRNA-seq offer a unique opportunity to explore and characterize neural cell organization across different cortical regions. Here, we mapped 14 human cortical regions, leading to a single-cell atlas comprising transcriptomic data from 1,121,772 nuclei and spatial profiles from 1,888,306 cells. The atlas reveals distinct expression patterns and spatial arrangements of neural cell types. Glutamatergic neurons show precise laminar patterns, with similar expression in adjacent areas. SST neurons fall into two transcriptional categories, corresponding to superficial and deep layer distributions. The atlas, integrated with functional networks, highlights correlations between neural cell types and cortical functions, uncovering cell-cell interactions and ligand-receptor patterns with regional differences in neuron-glia communication. It also deciphers transcriptomic differences and cellular composition in layer 4 and the stable subplate (layer 6b) across regions. Our findings offer insights into the cellular foundations of complex and intelligent regions within the human cortex.
Songren Wei,Meng Luo,Jiangping Xu,Rui Chen,Qinghua Jiang

4Spatial transcriptomics: The effect of consecutive slices data integration on accurate cell type annotation and clustering
(ID: STDS0000224)
In 10X Genomics Visium Spatial Gene Expression (ST), the resolution for distinguishing neighboring cells can be improved using data integration with single-cell/single-nuclei transcriptomics profiles. Besides, depending on the cell type and tissue, nuclei size may vary significantly to an extent that it may exceed the thickness of tissue slices. This may jeopardize capturing full transcriptomics profile of single slice due to the improper/incomplete incision of nuclei during cryosectioning process and this may cause drawbacks in downstream analysis. To monitor the probable consequences, we monitored the effect of consecutive slices data integration (CSDI) on improvement of cell type clustering and annotation through transferring cell labels from a single-nuclei transcriptomics dataset to ST. To do so, two consecutive slices from the orbitofrontal neocortex and temporal neocortex of two post mortem brain samples were obtained and their spatial transcriptomics profiles were retrieved using 10x Genomics Visium Spatial Gene Expression protocol. Using CSDI, not only the number of identified clusters were increased and the inconsistency between the pattern of clusters in consecutive slices was resolved, but the layered-structure of gray matter was unveiled. Besides, only after CSDI the transferred annotations from single-nuclei transcriptomics to ST could match the microscopic results. CSDI can improve the ST clustering and cell type annotation by providing the full signals coming from all cell types of single slice of tissue. The codes in R programming language are publicly available at https://github.com/ElyasMo/ST_snRNA-seq
Mieczkowski, Jakub,Mohammadi, Elyas,Chojnowska, Katarzyna,Bieńkowski, Michał,Kostecka, Anna,Magdalen
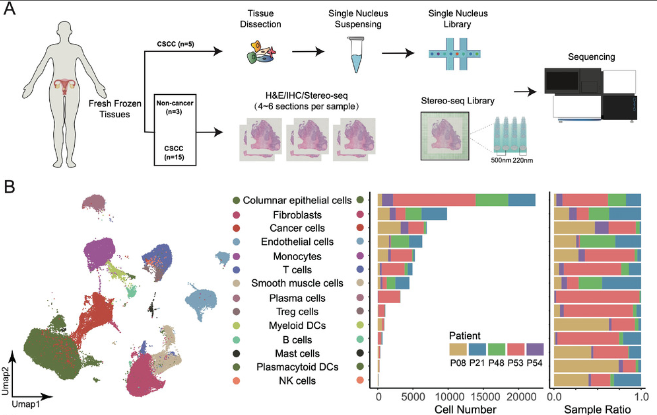
To understand the etiological, structural, and immunological characteristics of cervical squamous cell carcinoma (CSCC), we conducted spatial transcriptomics (ST) experiments for cervical samples from 16 individuals.
Xun Xu,Yanzhou Wang,Junhua Li,Peng Wu
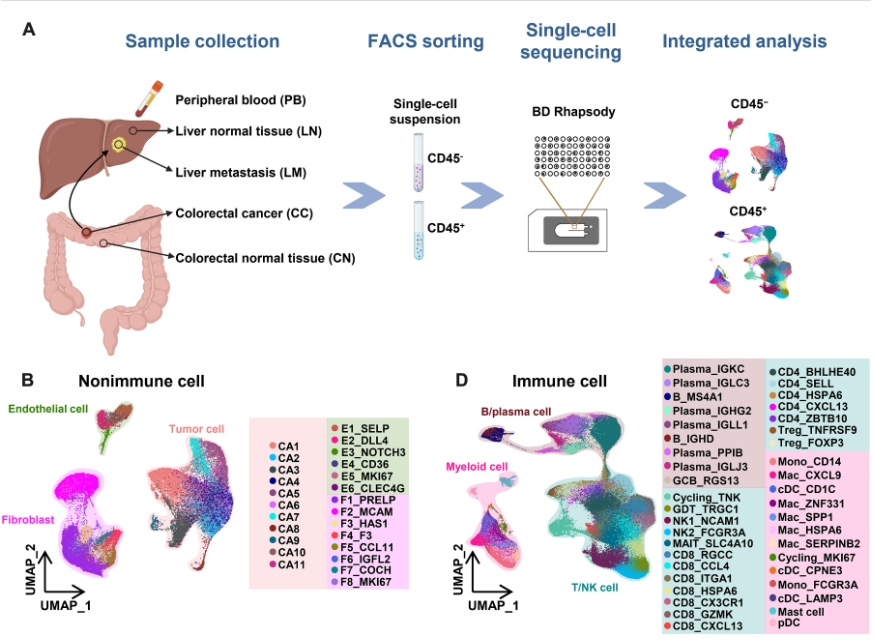
6Single-cell and spatial transcriptome analysis reveals the cellular heterogeneity of liver metastatic colorectal cancer
(ID: STDS0000216)
In this study, we comprehensively charted the cellular landscape of colorectal cancer (CRC) and well-matched liver metastatic CRC using single-cell and spatial transcriptome RNA sequencing. We generated 41892 CD45- non-immune cells and 196473 CD45+ immune cells from 27 samples of 6 CRC patients, and found that CD8_CXCL13 and CD4_CXCL13 subsets increased significantly in liver metastatic samples that exhibited high proliferation ability and tumor-activating characterization, contributing to better prognosis of patients. Distinct fibroblast profiles were observed in primary and liver metastatic tumors. The F3+ fibroblasts enriched in primary tumors contributed to worse overall survival by expressing pro-tumor factors. However, the MCAM+ fibroblasts enriched in liver metastatic tumors might promote generation of CD8_CXCL13 cells through Notch signaling. In summary, we extensively analyzed the transcriptional differences of cell atlas between primary and liver metastatic tumors of CRC by single-cell and spatial transcriptome RNA sequencing, providing different dimensions of the development of liver metastasis in CRC
Long; Jie; Wang; Fei

7Spatial resolution of cellular senescence dynamics in colorectal liver metastasis
(ID: STDS0000219)
Heterogeneity of senescent cancer cells have been dissected by Spatial Transcriptomic in human colorectal liver metastasis (CRLM).
Faggioli, Francesca,Lambroia, Luca

8Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment
(ID: STDS0000153)
Transcriptional heterogeneity among malignant cells of a tumor has been studied in individual cancer types and shown to be organized into cancer cell states; however, it remains unclear to what extent these states span tumor types, constituting general features of cancer. Here, we perform a pan-cancer single-cell RNA-Seq analysis across 15 cancer types and identify a catalog of gene modules whose expression defines recurrent cancer cell states including ‘stress’, ‘interferon response’, ‘epithelial-mesenchymal transition’, ‘metal response’, ‘basal’ and ‘ciliated’. Spatial transcriptomic analysis linked the interferon response in cancer cells to T cells and macrophages in the tumor microenvironment. Using mouse models, we further found that induction of the interferon response module varies by tumor location and is diminished upon elimination of lymphocytes. Our work provides a framework for studying how cancer cell states interact with the tumor microenvironment to form organized systems capable of immune evasion, drug resistance, and metastasis.
Barkley, Dalia,Yanai, Itai

9Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma
(ID: STDS0000001)
To define the cellular composition and spatial architecture of the tumor micoenvironment, we combined single-cell RNA-sequencing, spatial transcriptomics, and multiplexed ion-beam imaging from 10 patient cutaneous squamous cell carcinoma tumors and site-matched normal skin
Andrew Ji

5 µm section from Invasive Ductal Carcinoma of Homo sapiens Breast. FFPE tissue purchased from BioIVT Asterand Homo sapiens Tissue Specimens (BioIVT):