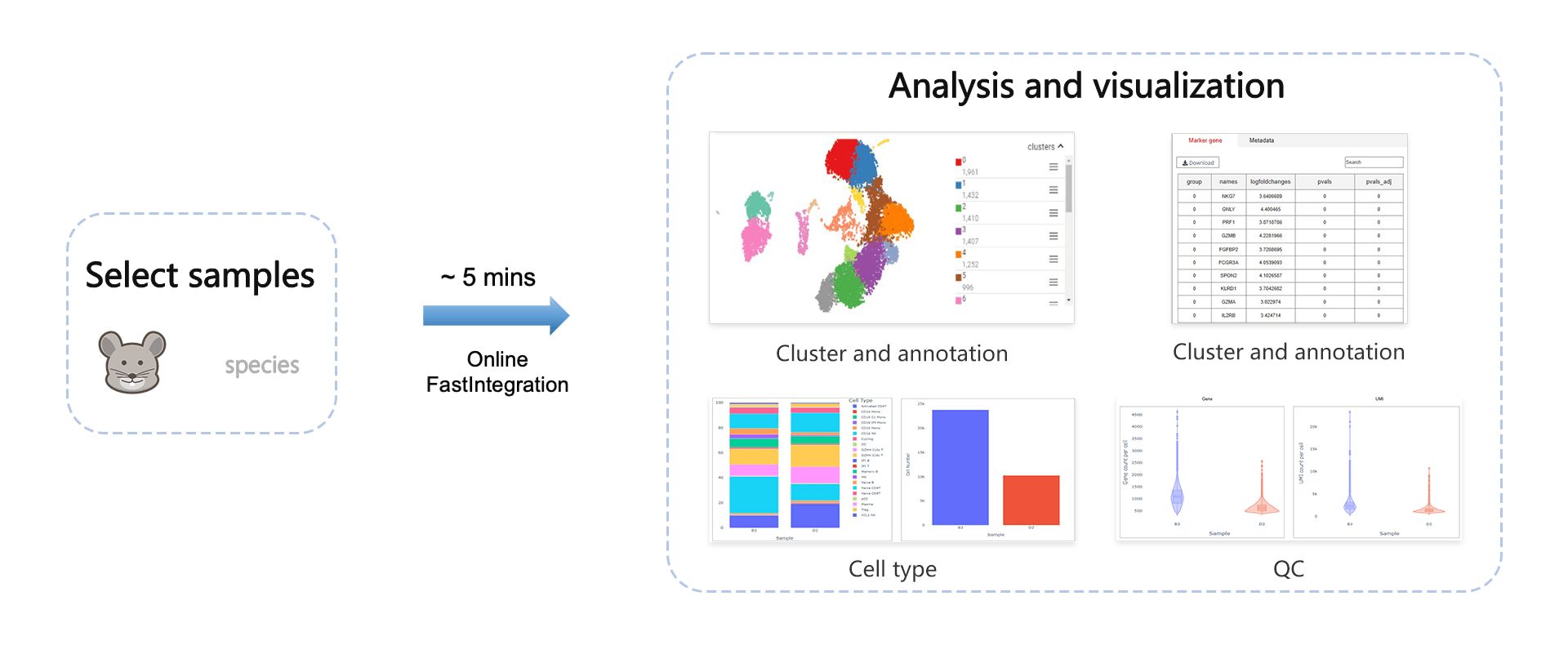
At CDCP (Cell-omics Data Coordinate Platform), we've introduced a powerful single-cell multi-sample joint analysis feature, making the exploration and interpretation of single-cell data more straightforward and intuitive. We're dedicated to providing a one-stop data collaborative analysis tool for researchers and analysts in various fields.
SingleR is an automatic annotation method for single-cell RNA sequencing (scRNAseq) data (Aran et al. 2019). Given a reference dataset of samples (single-cell or bulk) with known labels, it labels new cells from a test dataset based on similarity to the reference. Thus, the burden of manually interpreting clusters and defining marker genes only has to be done once, for the reference dataset, and this biological knowledge can be propagated to new datasets in an automated manner.

scMCA is a tool developed by Guo Guoji’s team based on single-cell gene expression to define mouse cell types. The basic computational approach involves constructing a reference system by averaging the expression values of 100 randomly sampled cells from known cell populations. The Pearson correlation coefficient is then calculated between the expression values of the test cell and the reference cell populations, and the cell population with the highest correlation is used as the basis for classification.
Tangram is a Python package based on scanpy, for mapping single-cell (or single-nucleus) gene expression data onto spatial gene expression data.
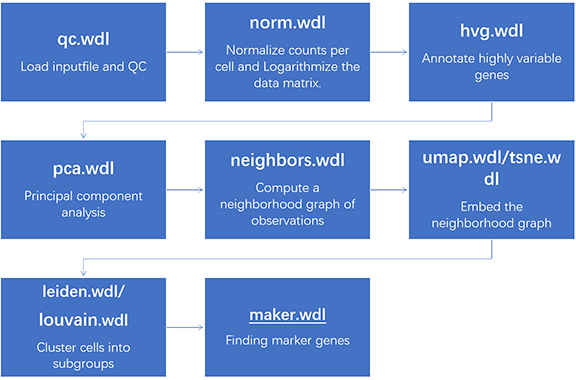
Scanpy is a commonly used and mainstream Python analysis toolkit for single-cell transcriptomics, used to analyze single-cell gene expression data combined with anndata. It includes preprocessing, visualization, clustering, trajectory inference, and differential expression testing. It can effectively handle datasets with over one million cells.