Subprojects
Fish phylogeny: the phylogenetic tree of all fishes
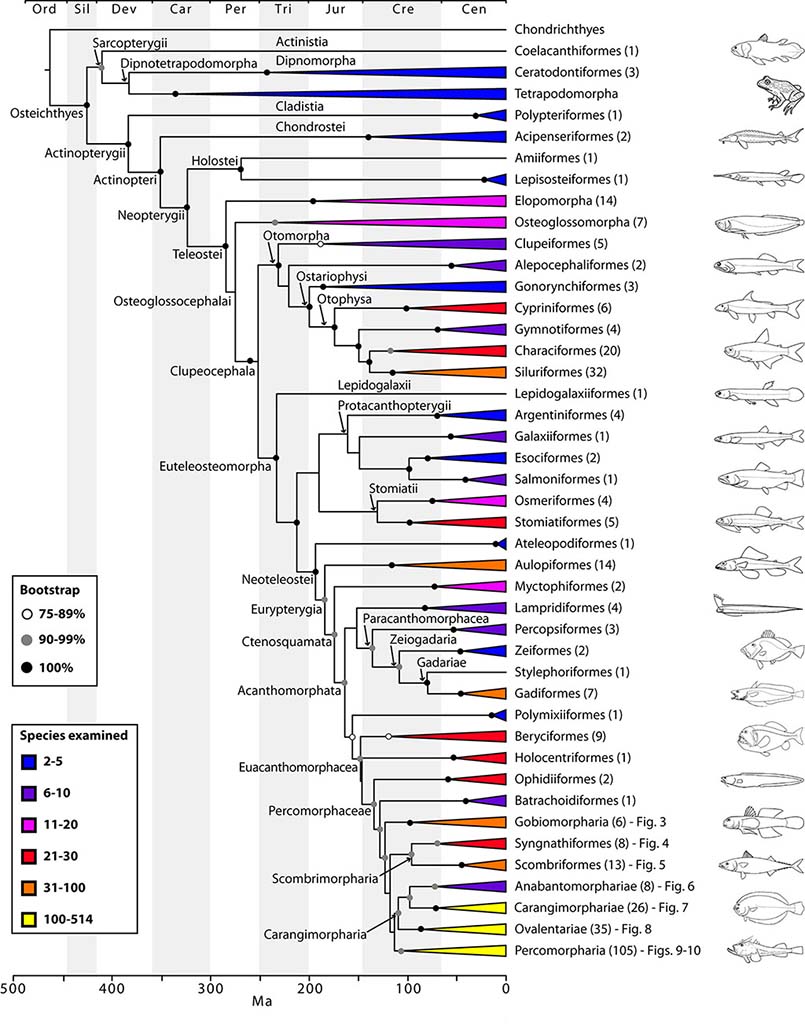
Ricardo Betancur-R, et al. 2013
Phylogenetic relationships among fishes are gradually being resolved with new sources of genetic data. Increasing resolution for the tree of life of fishes is emerging from recent studies using a modest number (10 to 20) of molecular markers. These studies provided unprecedented resolution and high support for most backbone nodes, defining for the first time a global phylogeny of all fishes. The general structure of this tree is in agreement with previous morphological and molecular studies, but unanticipated new clades have been proposed. The most significant result has been resolution of the historically recalcitrant uncertainty concerning affinities among the crown-group percomorph fishes, a group that contains about one third of all fish species (with about 30 orders and almost 300 families). The new molecular phylogenies resolved percomorph affinities into nine well-supported supraordinal clades, suggesting that their extant diversity has originated as a result of relatively recent independent radiations. However, poor phylogenetic resolution within the newly discovered percomorph clades hinders our understanding of the major factors affecting rates of morphological and lineage diversification. Future research should provide such necessary phylogenetic resolution to improve our ability to understand the evolution of the richest coral reef fish clades and the most mysterious deep-sea groups.
Several parts of the fish tree that require additional study include:
- the basal divergence of euteleosteomorph groups and circumscription of Protacanthopterygii, in particular interrelationships of argentiniforms, galaxiiforms, osmeriforms, salmoniforms, esociforms, stomiatiforms and neoteleosts;
- interrelationships among components within the new percomorph series Pelagimorpharia, Carangimorpharia, Ovalentaria, Percomorpharia, and the order Perciformes;
- the ordinal status of 55 percomorph families examined (as well as many others unexamined) that remain with uncertain ordinal affiliation (incertae sedis).
The Fish-T1K project will significantly contribute to the reconstruction of a comprehensive molecular phylogeny for all fish species to further resolve and test existing phylogenetic hypotheses and to provide better understanding of the evolution of the incredible diversity of fishes.
Coordination
- Guillermo Ortí (gorti@gwu.edu), Department of Biological Sciences, The George Washington University, USA.
- Byrappa Venkatesh (mcbbv@imcb.a-star.edu.sg), Institute of Molecular and Cell Biology, A*STAR, Singapore.
- Ying Sun (sunying@genomics.cn), Marine Biobank, BGI-Shenzhen, China.
- Qiong Shi (shiqiong@genomics.cn), State Key Laboratory of Agricultural Genomics, BGI-Shenzhen, China.
Evolutionary genomics of fish venoms








Venom, a complex biochemical cocktail of biologically active components, such as proteins, peptides, amino acids, neurotransmitters and polyamines, has underpinned the successful diversification of venomous animals, such as scorpions, spiders, and lionfish. Current research focusing on potential pharmacological agents or physiological tools found in the toxins of venomous animals (e.g. snakes) revealed several pharmaceuticals with function ranging from anticoagulation to antiangiogenesis.
Venomous fishes have been largely ignored as a source of potential pharmaceuticals compared to terrestrial animal groups, with only around one dozen toxins characterized to date from venomous fishes. However, recent findings suggest that there may be more venomous fish species than all other venomous vertebrates combined, yet little is known about the composition of venoms present in fishes. Therefore, venomous fishes represent an untapped resource of natural products with potential pharmaceutical benefits waiting for exploitation.
Thus, we will study representatives of venomous osteichthyes from Siluriformes, Beryciformes, Perciformes and Scorpaeniformes, using a combination of transcriptomics and peptidomics (LC-MS/MS sequencing), followed by extended bioinformatics analyses, to successfully identify the toxin-like peptides in fish venoms.
Coordination
- Agostinho Antunes(aantunes@ciimar.up.pt), CIIMAR, University of Porto, Portugal.
Microbes and marine-to-freshwater transitions in ray-finned fishes
Actinopterygiian fish inhabit an array of waters from the deep ocean, to lakes, rivers, and even seasonal ponds. The diversity of freshwater fish has been of particular interest to evolutionary biologists, as roughly half of all fish species inhabit the tiny fraction of aquatic environment that is freshwater. While ray-finned fish have invaded freshwater multiple times in their evolutionary history, the ability to cross this barrier is not evenly distributed, and many lineages are entirely marine.
The ability to invade freshwater is a complex trait. Changes in osmotic pressure are a challenge to many organisms, and a marine organism invading a freshwater environment will be forced to cope with that challenge (Lee and Bell, 1999). Teleost fishes are hypotonic to marine water, but hypertonic to freshwater, thus a marine fish must excrete excess salts taken up from seawater, and a freshwater fish will take in salts from the environment, to maintain homeostasis (Evans, 2008). A second immunological barrier has more recently come to light through metagenomic studies of salt and freshwater environments. Viruses and bacteria have made the marine-freshwater transition relatively few times in their evolutionary history (Logares et al., 2009), and salinity is the strongest determinant of microbial community structure (Lozupone and Knight, 2007). Given these differences, the immune system of fishes should play a very significant role in the adaptation to freshwater, but this role has not been well investigated.
No organism is an island, and we are becoming increasingly aware of the constant interactions between microbes and their vertebrate hosts. Early studies of fish microbiomes tended to reflect only the bacterial community that could be detected through culture techniques, but advances in sequencing technology allow researchers to characterize the microflora more completely (Austin, 2006). The microbiome of aquacultured fishes has been of interest, though studies of wild fish populations have been rare (Sevellec et al., 2014). Most research has focused on establishing the teleost gut microbiome (Llewellyn et al., 2014), but even this area research is far behind that on other vertebrates (Clements et al., 2014).
When fish transition between marine and freshwater environments, it is likely that they are encountering an entirely new spectrum of microbes, and this may significantly change their microbiome. Gills secrete mucus that contains lysozyme and other anti-microbial agents as a first defense against pathogens (Cahill, 1990; Gomez et al., 2013; Press and Evensen, 1999). Even so, bacteria are often found in large quantities on gill tissue, and both commensal and pathogenic species can inhabit the mucosal layer (Austin, 2006; Cahill, 1990; Gomez et al., 2013). There is some evidence that the bacterial flora of fish is heavily influenced by the environment, with evidence of differences across latitude and temperature (Cahill, 1990), as well as differences associated with salinity in the same species (Horsley, 1973). Given the strong relationship between bacterial community structure and salinity (Lozupone and Knight, 2007), it is unsurprising that documented differences in fish microbiota have been correlated with salinity gradients. The functional profile of these bacteria is not known in most cases, except where pathogens have been identified.
The Fish-T1K project offers a unique opportunity to survey the microbiota of fishes across the fish tree of life, and to gain insight into how salinity has shaped differences at a broad scale. Additionally, we can ask questions about how the different microbial communities between marine and freshwaters have shaped the underlying genetic basis of the immune systems of the fish that inhabit these different environments. The focus on gill tissue is ideal for such a study, given it’s immunological function and its direct interactive with the environment.
We would like to propose using the following pairs of already sequenced species: Amblygaster clupeoides and Misgurnus anguillicaudatus (Otomorpha); Liza affinis and Glossolepis incisus (Ovalentaria); Coreoperca whiteheadi and Amblycirrhitus pinos (Percomorpharia). Using pairs of marine and freshwater species sequenced by the Fish-T1K project, we can survey microbes present in the gill using Pathoscope 2.0, which uses a likelihood model to assign a genomic origin of a particular sequencing read based on a genome database(Hong et al., 2014). This will allow us to ask whether there are significant differences in the microbiome community of marine and freshwater fishes, but also identify whether the functional profile of those microbiomes is significantly different across environments. It will also allow us to assess whether environment or phylogenetic relatedness is more important in shaping the microbial community of the gill.
Further armed with transcriptome data, we can investigate whether differences in microbial communities has resulted in a different selective regime on the immune systems of marine and freshwater fish. By identifying orthologous transcripts of genes related to immune functions across our study species, we can test hypotheses about the nature of the selection on immune systems between these two very different environments. Using nonsynonymous -to-synonymous substitution rates to detect positively selected genes and testing for increased rates of evolution (fast-evolving genes, sensu (Backstrom et al., 2013)), we can test the hypothesis that the different microbial community in freshwater results in a different selective regime on the immune system of fishes. A significantly increased number of positively selected and fast-evolving immune genes would support the idea that the immune system is important in allowing fish to adapt to freshwater.
References
- Austin, B., 2006. The bacterial microflora of fish, revised. The Scientific World Journal 6, 931-945.
- Backstrom, N., Zhang, Q., Edwards, S.V., 2013. Evidence from a house finch (Haemorhous mexicanus) spleen transcriptome for adaptive evolution and biased gene conversion in passerine birds. Molecular biology and evolution 30, 1046-1050.
- Cahill, M.M., 1990. Bacterial flora of fishes: a review. Microbial ecology 19, 21-41.
- Clements, K.D., Angert, E.R., Montgomery, W.L., Choat, J.H., 2014. Intestinal microbiota in fishes: what's known and what's not. Molecular ecology 23, 1891-1898.
- Evans, D.H., 2008. Teleost fish osmoregulation: what have we learned since August Krogh, Homer Smith, and Ancel Keys. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 295, R704-R713.
- Gomez, D., Sunyer, J.O., Salinas, I., 2013. The mucosal immune system of fish: the evolution of tolerating commensals while fighting pathogens. Fish Shellfish Immunol 35, 1729-1739.
- Hong, C., Manimaran, S., Shen, Y., Perez-Rogers, J.F., Byrd, A.L., Castro-Nallar, E., Crandall, K.A., Johnson, W.E., 2014. PathoScope 2.0: a complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2, 1-15.
- Horsley, R.W., 1973. The Bacterial Flora of the Atlantic Salmon (Salmo salar L.) in Relation to its Environment. Journal of Applied Bacteriology 36, 377-386.
- Lee, C.E., Bell, M.A., 1999. Causes and consequences of recent freshwater invasions by saltwater animals. Trends in Ecology & Evolution 14, 284-288.
- Llewellyn, M.S., Boutin, S., Hoseinifar, S.H., Derome, N., 2014. Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Frontiers in microbiology 5, 207.
- Logares, R., Brate, J., Bertilsson, S., Clasen, J.L., Shalchian-Tabrizi, K., Rengefors, K., 2009. Infrequent marine-freshwater transitions in the microbial world. Trends in microbiology 17, 414-422.
- Lozupone, C., Knight, R., 2007. Global patterns in bacterial diversity. Proceedings of the National Academy of Sciences of the United States of America 104, 11436-11440.
- Press, C.M., Evensen, Ø., 1999. The morphology of the immune system in teleost fishes. Fish & Shellfish Immunology 9, 309-318.
- Sevellec, M., Pavey, S.A., Boutin, S., Filteau, M., Derome, N., Bernatchez, L., 2014. Microbiome investigation in the ecological speciation context of lake whitefish (Coregonus clupeaformis) using next-generation sequencing. Journal of evolutionary biology 27, 1029-1046.
Coordination
- Lily Hughes (lilychughes@gwu.edu), Department of Biological Sciences, The George Washington University, USA.
- Guillermo Ortí (gorti@gwu.edu), Department of Biological Sciences, The George Washington University, USA.
- Keith Crandall (kcrandall@gwu.edu), Computational Biology Institute, The George Washington University, USA.