Introduction of home and data page
Introduction of home page
1. Filter your job that running on DISSECT
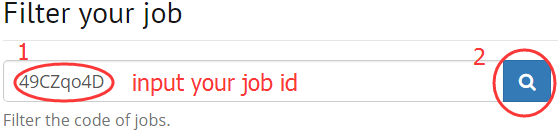
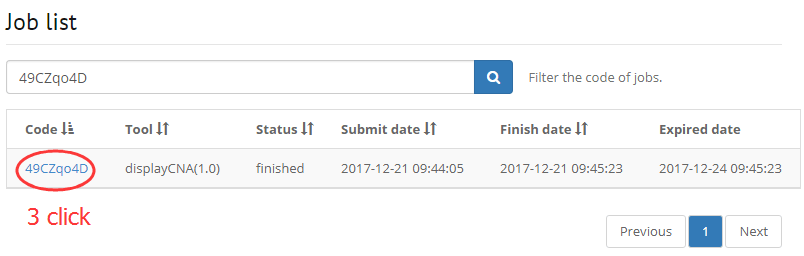
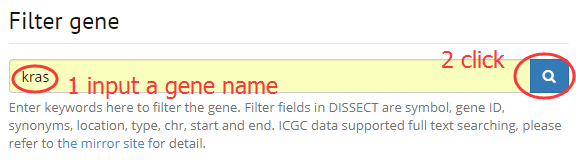
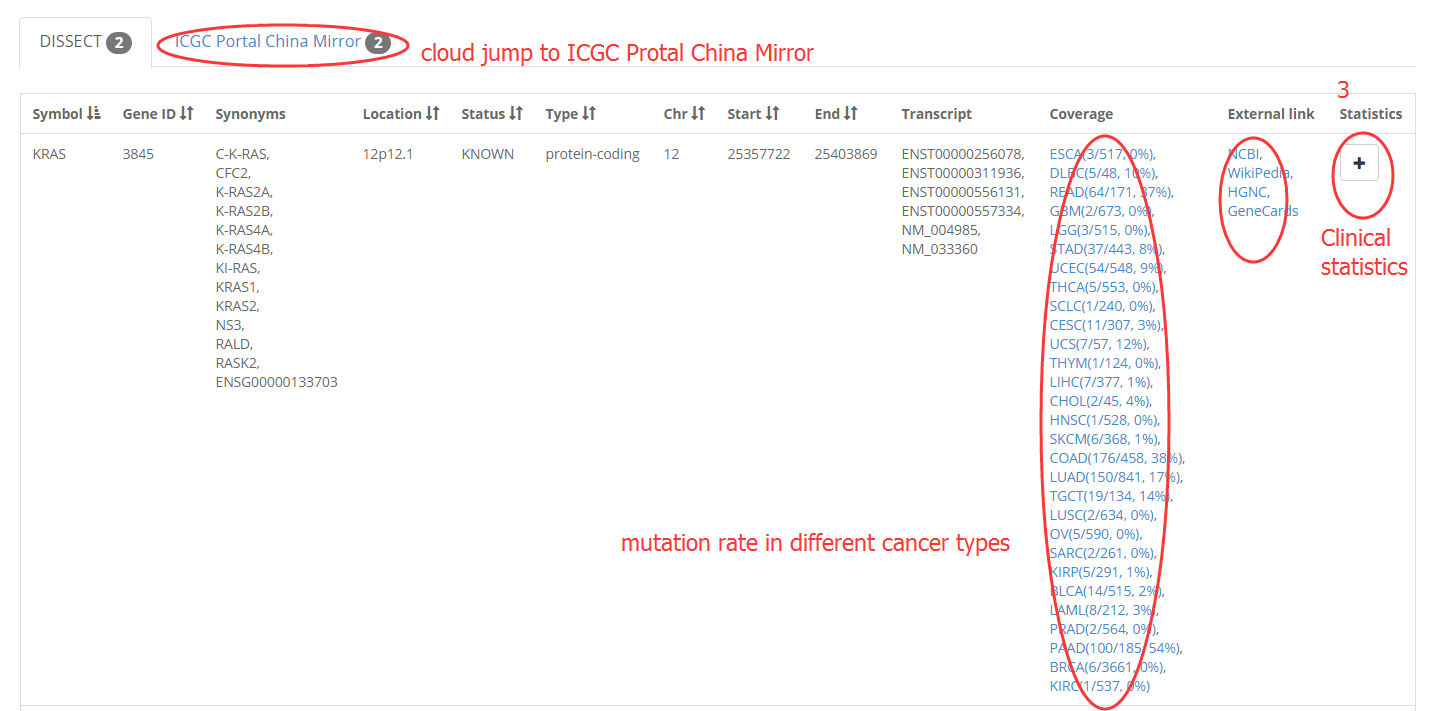
2. Filter a gene by input symbol, gene ID, synonyms.
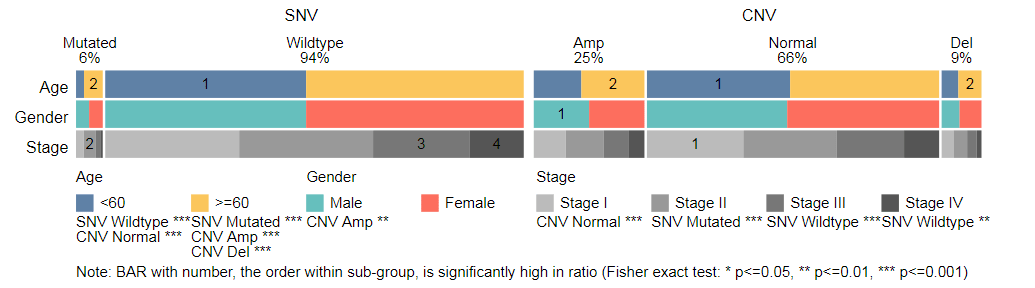
Introduction of data page
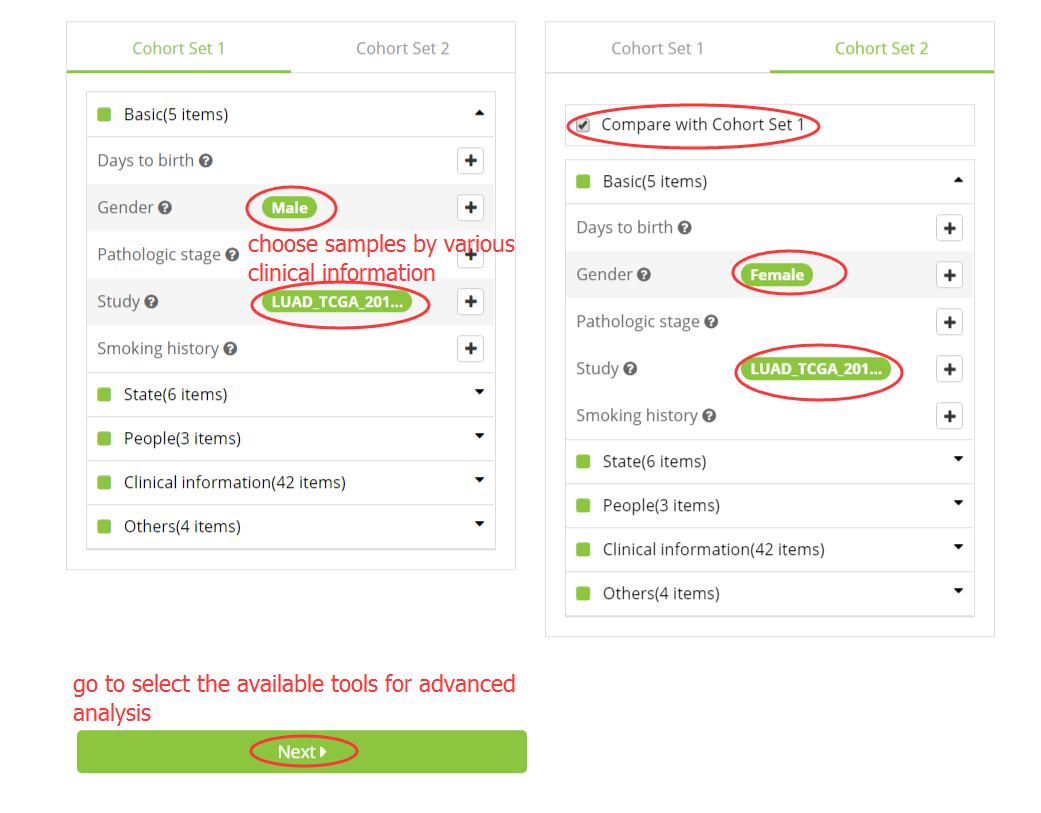
1. Select samples for analysis
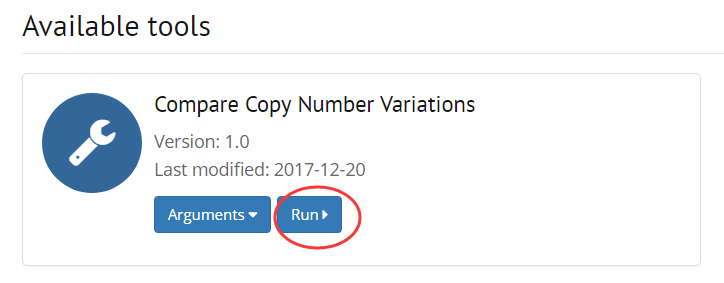
2. Choose an available tool for the selected samples
ACMG help
Introduction of Variants Classifier
The American College of Medical Genetics and Genomics (ACMG) published in 2015 the updated standards and guidelines for the clinical interpretation of sequence variants, based on 28 criteria. However, there are still extensive discrepancies among different laboratories.
Variants Classifier is a software tool based on ACMG guideline, aiming to aid clinical explanation of germline variants in genes related with cancer. The input to Variant Classifier is set to be one of four formats: 1 genomic coordinate (Chromosome:Position:Ref:Alt), e.g. 1:155209747:A>C; 2 dbSNP ID, e.g. rs730881285 ; 3 cDNA change, e.g. ATM: NM_000051,c.T6122C; 4 VCF file, and the output is the ACMG classification and detailed explanation of evidences .
Instructions
1 Upload your variant with one of the following four formats:
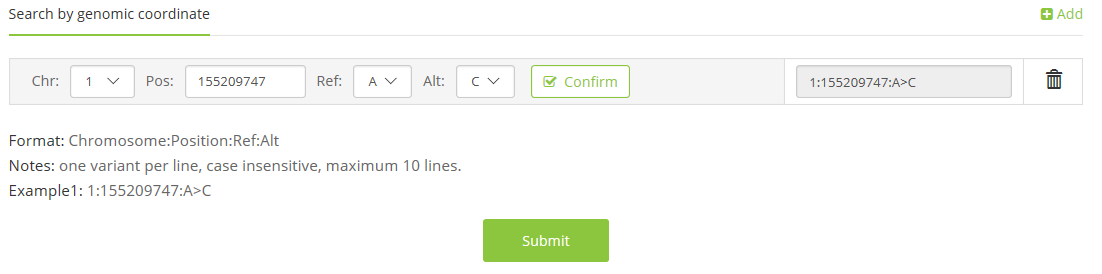
1.1 coordinate on reference genome(Chromosome:Pos:Ref>Alt),
e.g. 1:155209747:A>C;
1.2 dbSNP ID,
e.g. rs730881285;
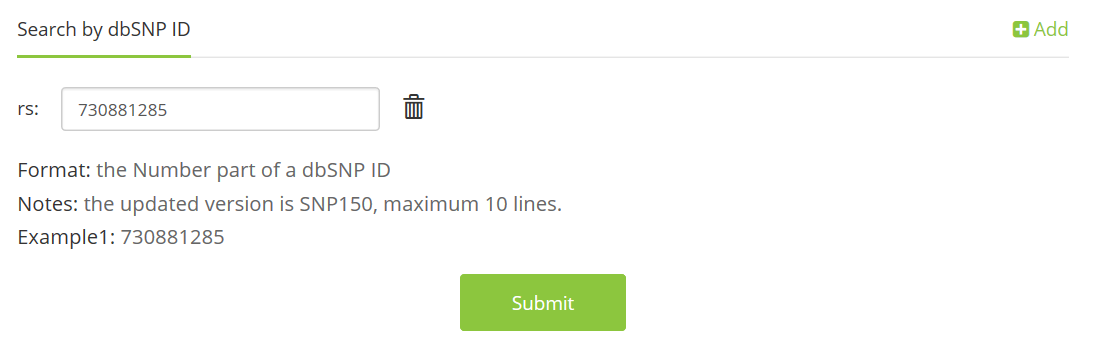
1.3 cDNA change,
e.g. ATM: NM_000051,c.T6122C;
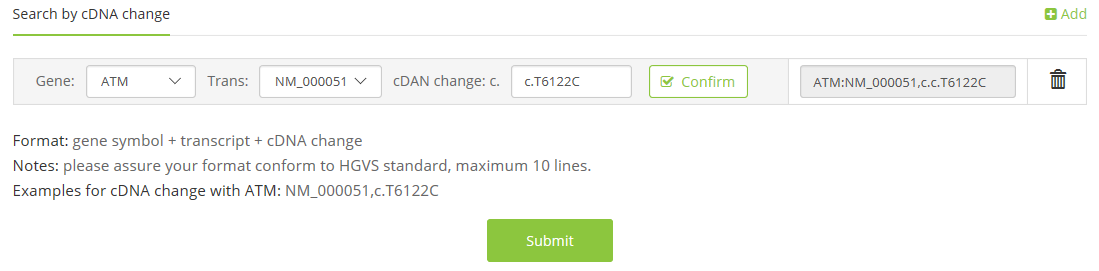
1.4 VCF file,
e.g. test.vcf,
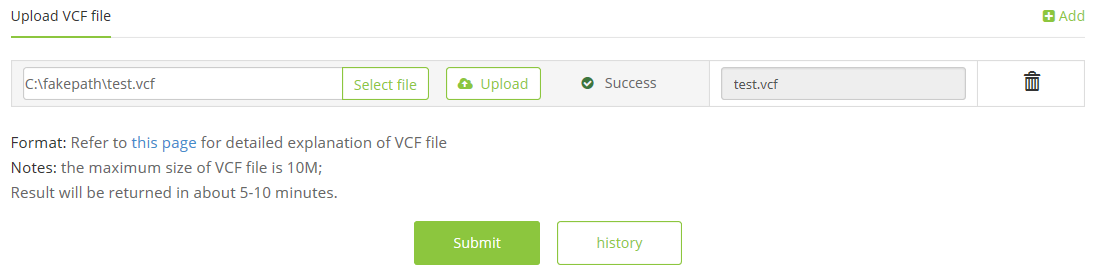
2 Click the ‘Submit’ button
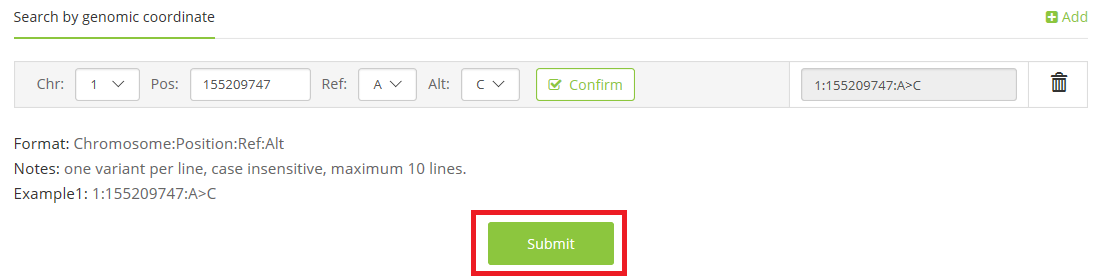
3 waiting for seconds or minutes (if you choose to upload a VCF file), and the output will be shown with class, evidence level, and detail explanation
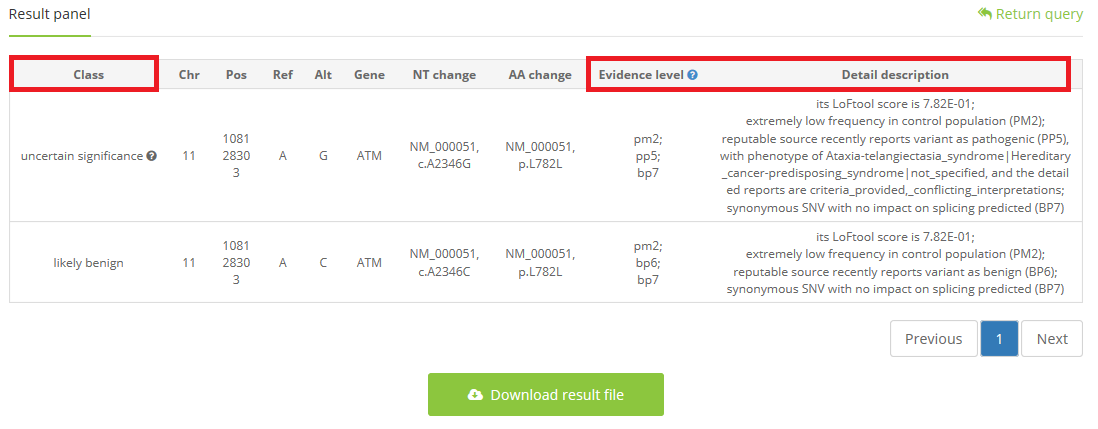
A blue question markwill be shown on the upper right of the ‘Evidence level’, and move your cursor there, a general instruction for varieties of evidence will be shown automatically.
When classification is ‘uncertain significance’, a gray question mark will exist, and move your cursor there, artificial tips will be shown automatically which can help clinical expert to search on other databases for supporting evidence to make a final judgment.
ACMG evidence level
- Pvs1: wholegene deletion, stopgain, frameshift, or splicing mutation in 2bp
- Ps1: same amino acid change as known pathogenic variants, with different nucleic acid alteration
- Pm2: no record OR lower than 0.0003 in 1000g2015aug(ALL,EAS), exac03(ALL,_EAS)
- Pm4: in-frame deletion/insertion OR stop codon loss which resulted the length change of translated protein
- Pm5: same site as the known pathogenic variant with different nucleic acid change
- Pp3: multiple lines of computational evidence support a deleterious effect on the gene or gene product
- Pp5: reputable source recently reports variant as pathogenic
- Pp6: ocated in the hotspot area or specific function domain
- Ba1: Allele frequency is >1%
- Bs1: Allele frequency is greater than expected for disorder
- Bp1: missense variant while most pathogenic variants are truncating variants
- Bp3: in-frame deletions/insertions in a repetitive region without a known function
- Bp4: multiple lines of computational evidence suggest no impact on gene or gene product
- Bp6: reputable source recently reports variant as benign
- Bp7: a synonymous variant for which splicing prediction algorithms predict no impact to the splice consensus sequence