Generation of Stereo-seq chip.
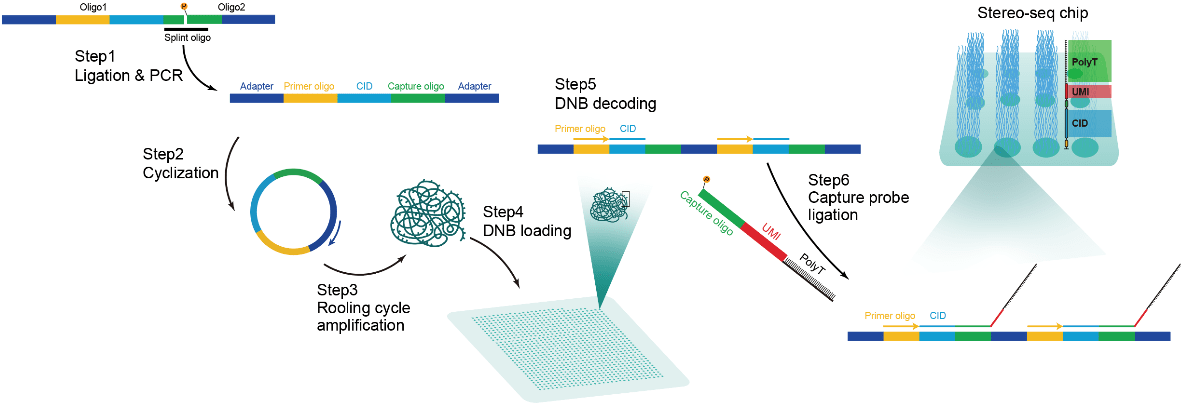
To generate the patterned array, we first synthesized two oligo sequences: one containing 25 random deoxynucleotides ( TGTGAGCCAAGGAGTTGNAACTGCTGACGTACTGAGAGGCATGGCGACCTTATCAGNNNNNNNNNNNNNNNNNNNNNNNNNTTGTCTTCCTAAGACCG , Sangon) and the other a fixed sequence with phosphorylation ( /5phos/CTTGGCCTCCGACTTAAGTCGGATCGTAGCCATGTCGTTC, Sangon). These two oligos were ligated by incubation with T4 ligase (NEB; 1U/μl T4 DNA ligase and 1× T4 DNA ligation buffer) and splint oligo ( TCGGAGGCCAAGCGGTCTTAGGAA, Sangon, 1 μM) at 37℃ for 2 hours. The products were purified using the Ampure XP Beads (Vazyme, N411-03) and then PCR amplified with the following steps: 95℃ for 5 minutes, 12 cycles at 98℃ for 20 seconds, 58℃ for 20 seconds, 72℃ for 20 seconds and a final incubation at 72℃ for 5 minutes. The PCR products were purified using the Ampure XP Beads (Vazyme, N411-03). DNBs were then generated by rolling circle amplification and loaded onto the patterned chips according to the MGI DNBSEQ-Tx sequencer manual. Next, to determine the distinct DNB-CID sequences at each spatial location, single-end sequencing was performed using a sequencing primer ( CTGCTGACGTACTGAGAGGCATGGCGACCTTATCAG, Sangon, 1 μM) in a MGI DNBSEQ-Tx sequencer with SE25 sequencing strategy. After sequencing, the capture oligo ( /5phos/TTGTCTTCCTAAGACNNNNNNNNNNTTTTTTTTTTTTTTTTTTTTTV, Sangon, 1 μM) including 22 nt poly-T and 10 nt UMI was hybridized with the DNBs in 5× SSC buffer at 37℃ for 30 minutes, and then incubated with T4 ligase (NEB, 1 U/μl T4 DNA ligase, 1× T4 DNA ligation buffer and 0.5% PEG2000) at 37℃ for 1 hour. This procedure produces capture probes containing a 25 nt CID barcode, a 10 nt UMI and a 22 nt poly-T ready for poly-A RNA capture.
Stereo-seq library preparation.
-
The Integral Processes of Stereo-seq
- Specimen Preparation and Embedment
- Cryosection
- RNA Quality Assessment
- Chip Treatment and Section Placement
- Tissue Fixation and Imaging
- Tisse Permeabilization
- Reverse Transcription
- Tissue Removal
- RT Product Collection and Amplification
- cDNA Purification
- cDNA Fragmentation and Amplification
- Size Selection of Fragmentation Product
-
Flowchart

-
Detailed Procedures of Stereo-seq
-
Take a fresh or liquid nitrogen fresh-frozen tissue. Two methods are provided for tissue preparation—A. Fresh Tissue embedment and B. Liquid Nitrogen Fresh-Frozen Tissue Embedment. The former is recommended.
-
Fresh Tissue Embedment
- Fill two-thirds of a metal beaker with isopentane (sufficient to fully submerge the tissue) and place in a liquid nitrogen dewar (same level as isopentane) to allow sufficient contact.
- Incubate 15 minutes. Pre-cool 1× PBS at 4℃ and OCT on ice for 10 minutes.
- Pour a layer of pre-cooled OCT, about 3 mm in thickness, into the tissue processing embedding cassette. Place the cassette into 250 ml beaker above to thoroughly solidify OCT. Keep the OCT surface flat.
- Clean a fresh tissue with cooled 1× PBS twice. Dry it out with Kimwipes. For frozen tissue, skip to step 5.
- Be aware of the embedding direction. Place the fresh/frozen tissue on the solid OCT in the cassette by tweezers and adjust to an optimum direction for cryosection. Complete sample embedding by covering the sample with prechilled OCT, avoiding bubbles, especially around the tissue.
- Pick up the cassette within the embedding tissue and hold it into the prechilled isopentane until the OCT is completely solid.
- Mark the embedding direction of tissue. Keep the embedded tissue in a -80℃ freezer until cryosection.
-
Liquid Nitrogen Fresh-Frozen Tissue
- Attention! No deformation on tissues.
- Clean the tissue with pre-cooled 1× PBS on ice for three times in a culture dish. If the tissue cannot be frozen immediately, it can be kept in pre-cooled 1× PBS at 4℃ for less than 1 hour. Liquid nitrogen fresh-freezing should be completed within 1 hour.
- Use Kimwipes to remove the liquid. Avoid moisture residue crystallizing on the tissue surface, which could affect embedding and cryosection followed.
- Transfer the tissue into a proper size of tube without squeezing. The tissue will attach the tube wall automatedly.
- Insert the tube horizontally into the liquid nitrogen for 10 minutes then keep the sample tube in a -80℃ freezer for Stereo-seq, 3 years at most.
- Embed the tissue following Method A Step 5 ~ 7.
-
-
Cryosections
- Pre-cool the cryochamber. Set the chamber temperature to -20℃ and the microtome temperature to -10℃ ~ -15℃, depending on tissue type, status and environment temperature.
- Place tweezers, brushes and blades inside the chamber at -20℃ for pre-cooling.
- Equilibrium an embedded specimen in the chamber.
- Cut the excess OCT of the embedded specimen for appropriate size and direction.
- Place a cryostat chuck on top of the Quick-freeze shelf and activate cooling. Pour some pre-cooled OCT on the top of the shelf. Then adhere the embedded specimen to the OCT. Wait for OCT solidification
- Trim the section into the size fitting the chip and cryosection.
Caution: The temperature of the microtome should be in a proper range depending on the tissue type and the environment temperature. A temperature too high curls the sections and a temperature too low cracks the sections.
-
RNA Quality Assessment
To maintain the quality of RNA in the specimen, an assessment on RNA quality is suggested. Cut 10~20 pieces of 10 μm sections and put them into a -20℃ pre-cooled 1.5 ml Eppendorf tube. Isolate the RNA from the sections. For the assessment, RIN ≥ 7 is qualified. The remaining embedded specimens can be covered by a thin layer of OCT and wrapped in Parafilm or kept on the chuck in the cryochamber until cryosection after the assessment.
-
Chip Treatment and Cryosection
-
Transfer the stereo-seq chip to a new 24-well cell culture dish carefully. Record the original and
fraction numbers of the fluorescent chip.
Caution: The chips have front and back sides, KEEP the front side upwards, DO NOT upside down, DO NOT scratch the surface. - Wash the chip twice using 400 μl of 0.1× SSC then discard the surface liquid by pipetting.
- Add 100 μl of Nuclease-free water (NF-H2O) containing RNase Inhibitor (RI) to cover the chip surface and stay for 1~2 minutes, then discard the surface liquid by pipetting.
- Wash the chip by 400 μl of NF-H2O (without RI), discard the liquid.
- Transfer the chip to a clean Kimwipes by tweezers to dry the chip back and side.
-
Dry the chip at room temperature or on a Thermocycler Adaptor pre-set to 37℃ until no moisture left
on the chip surface,then keep the chip at a dry plate. DO NOT leave the chip at 37℃ over-time.
NF-H2O (with RI) 1× +10% (μl) NF-H2O 110 RI (RNase Inhibitor 40 U/μl) 5.5 Total 115.5 -
Cryosections and place the section on a chip carefully, then quickly dry the chip at 37℃ by
Thermocycler Adaptor for 3 minutes. Then move to the next step immediately.
Caution: Dry upon section placement. No stay at room temperature.
-
Transfer the stereo-seq chip to a new 24-well cell culture dish carefully. Record the original and
fraction numbers of the fluorescent chip.
-
Tissue Fixation and Imaging
- Immediately after drying, immerse the chip into pre-cooled methanol (-20℃) for fixation (30 ~ 40 minutes).
-
After fixation, take out the chip and wipe off the excess methanol on the back. Place the chip in
the parafilm-attached cell culture dish.
Tissue Fluorescence Staining Solution
Reagents 1× + 10%(μl) 5× SSC 220 InvitrogenTM Qubit ssDNA HS Reagent 1.1 RI (RNase Inhibitor 40 U/μl) 11 Total 232.1 - After methanol completely vaporized, drip 100 μl of tissue fluorescent staining solution on the chip. Stain for 15 minutes at room temperature, away from light.
- Remove the staining solution and wash the chip one time using 100 μl of 0.1× SSC (1/20 RNase Inhibitor contained). Keep moist.
- Exchange the 0.1× SSC by SRE (1/20 RNase Inhibitor contained). Take fluorescence image from the chip. Microscope setting: FITC channel, 10× objective lens, full scan on capture areas.
- Exchange the SRE by 100 μl of 0.1× SSC (1/20 RNase Inhibitor contained).
- Remove the liquid on the surface. Wipe off the excess liquid on the back and side then add permeabilization reagent immediately.
Caution: Drip solution slowly and do not wash away the tissue.
-
Tissue Permeabilization
-
Permeabilization reagent (prepare upon using) preparation: add 1 ml of freshly prepared 0.01N HCl to
10 μl of permeabilization enzyme. Pipette-mix thoroughly. Do not vortex. Incubate the as-prepared
permeabilization reagent at 37℃ for 3 minutes before use.
Caution: Do not vortex permeabilization enzyme. Pipette-mixing before use. -
Place the chip-contained cell culture dish in an incubator, heating to 37℃. Drip the pre-warmed
permeabilization reagent on to the chip drop by drop, 100 μl per chip.
Caution: Uniformly cover the tissue using permeabilization reagent. - Remove permeabilization reagent. Wash the chip using 100 μl of 0.1× SSC (1/20 RNase Inhibitor contained).
- Remove the liquid on the surface. Wipe off the excess liquid on the back and the side. Place the chip in a new cell culture dish. Add RT Mix immediately.
-
Permeabilization reagent (prepare upon using) preparation: add 1 ml of freshly prepared 0.01N HCl to
10 μl of permeabilization enzyme. Pipette-mix thoroughly. Do not vortex. Incubate the as-prepared
permeabilization reagent at 37℃ for 3 minutes before use.
-
Reverse Transcription
-
Immerse the chip into 400 μl of gently added RT Mix (pre-warmed to room temperature).
RT Mix
Reagents: 1× + 10% (μl) RT Reagent 352 RT Oligo 22 DTT (0.1 M) 22 RT Enzyme Mix 22 RNase Inhibitor 22 Total 440 - Reverse transcription: patch the chip-contained well by parafilm, incubated at 42℃ for 3 hours or overnight.
-
Immerse the chip into 400 μl of gently added RT Mix (pre-warmed to room temperature).
-
Tissue Removal
- Remove the RT Mix. Wash the chip by 0.1× SSC, 400 μl per well.
- Add 400 μl of tissue removal buffer per well and incubate at 37℃ for 30 minutes.
- Discard the tissue removal buffer. Wash the chip twice using 0.1× SSC, 400 μl per well. Transfer the washed chip to a new well for the next step.
-
Collection and Amplification of RT Product
-
Exo I digestion: Add Exo I Mix on to the chip, 400 μl per well, and incubated at 37℃ for 1 hour.
Exo I Mix
Reagents: 1× + 10% (μl) Exonuclease I 22 10× Exo I Reaction Buffer 44 H2O 374 Total 440 - Discard the liquid in the well. Wash the chip twice using 0.1× SSC, 400 μl per well.
- Wipe off the liquid on the back and the side of the chip.
- Place the chip on the Kimwipes. With the one hand fixing the edge of the chip from one side, crush the chip into pieces, normally 4 pieces, by the rear-end of tweezers using the other hand. Every two pieces are placed in one PCR tube, back-to-back.
-
Chip-PCR Mix preparation: Add 100 μl of Chip-PCR Mix to the tube to immerse the chip. Avoid air
bubble.
Caution: Make the front side of the chip fully contact with the Chip-PCR Mix.Chip-PCR MixReagents 1× + 10% (μl) H2O 46.2 cDNA HiFi Master Mix 55 cDNA Primers 8.8 Total 110 PCR Program Setting:Lid Temperature Reaction Volume Run Time 105℃ 100 Step Temperature Time 1 95℃ 00:05:00 2 98℃ 00:00:20 3 58℃ 00:00:20 4 72℃ 00:03:00 5 Go to Step 2,the recommended
Cycle number is 156 72℃ 00:05:00 7 12℃ Hold -
Run Qubit test on PCR products and record the concentrations (normally around 10~50 ng/μl).
Vortex the Qubit dsDNA Mix. For the test, mix 199 μl of the Qubit dsDNA Mix and 1 μl of the PCR product in a Qubit tube. Record the result.Qubit dsDNA Mix
Reagents: 1× + 10% (μl) InvitrogenTM Qubit dsDNA HS Buffer 218.9 Qubit dsDNA HS Reagent 200× 1.1 Total 220
-
Exo I digestion: Add Exo I Mix on to the chip, 400 μl per well, and incubated at 37℃ for 1 hour.
-
cDNA Purification: 0.6× Beads
- Warm the VAHTSTM DNA Clean Beads (VAZYME) at room temperature for at least 30 minutes. Pipette the Chip-PCR product to a new 1.5 ml EP tube and mix it with the beads in a ratio of 1:0.6. Vortex the mix then incubated at room temperature for 10 minutes.
- Spin down and place EP tubes on magnets for 3 minutes. Discard the supernatant while it is clean.
- Add 1 ml of newly prepared 80% ethanol and wait for 30 seconds.
- Discard the supernatant while it is clean. Repeat step 3 and 4 one more time.
- Air-dry the beads for 5-8 minutes until the beads surface is not reflective without cracking.
- Mix the dried beads with 42 μl of NF-H2O (or TE buffer). Vortex the mixture then placed on the magnets for 3-5 minutes. Collect the supernatant while it is clean. (You can stop at this step and store the sample at -20℃).
- Drip 1 μl of the purified cDNA and measure the concentration via Qubit dsDNA HS Kit. Detect fraction distribution of the cDNA via Agilent 2100 High Sensitivity Chip.
- QC Standard: Main peak locates at around 1500 bp.
-
cDNA Fragmentation and Amplification
Caution: Perform 3 groups of fragmentation on each sample and combine the products. Fragmentation enzyme is fragile to vortex. Pipette-mix while diluting. Ice-bath is required.
-
Fragmentation Mix:
Note: Input of cDNA product: X(ul)=(20 ng)/(Concentration of cDNA (ng/ul))
Reagents: 1× (μl) 5× TAG 4 10-fold NF-H2O-diluted Fragmentation Enzyme 0.5 cDNA Product X NF-H2O 15.5 - X Total 20
Prepare the fragmentation mix by gentle pipette-mixing. Avoid vortex. -
Thermal Cycler Program Setting:
Start the program above. Place the fragmentation mix-loaded PCR tube into the cycler while the temperature reaches 55℃. Set up a timer for 10 minutes upon closing the lid.
Lid Temperature Reaction Volume Run Time 75℃ 20 10minutes Step Temperature Time 1 55℃ 00:10:00 2 12℃ Hold - While time is up, add 5 μl of 5× NT Buffer to each tube with pipette-mixing. Place the tubes at room temperature for 5 minutes to terminate the reaction.
-
Amplification of the fragmented product: PCR amplification mix preparation
Reagents: 1× (μl) Fragmented Product Mix from the last step 25 Library HiFi Master Mix 50 Library PCR Primer Mix 25 Total 100 -
Vortex and spin down. Place the mix into thermal cycler following the program below.
Lid Temperature Reaction Volume Run Time 105℃ 100 Step Temperature Time 1 95℃ 00:05:00 2 98℃ 00:00:20 3 58℃ 00:00:20 4 72℃ 00:00:30 5 Go to Step 2,the recommended
Cycle number is 136 72℃ 00:05:00 7 12℃ Hold - Take 1 μl of product and measure its concentration via Qubit dsDNA HS Kit. Normally concentration is around 5~30 ng/μl. Record the result.
-
Fragmentation Mix:
-
Size Selection on Fragmentation Product (0.6× + 0.2×)
- Mix 100 μl of the PCR amplification product above with VAHTSTM DNA Clean Beads (VAZYME) that is pre-warmed to room temperature in a ratio of 1 : 0.6. Vortex then incubate at room temperature for 5 minutes.
- Spin-down and place the PCR tubes on magnets. After 3 minutes, transfer supernatant to a new tube while it is clear.
- Add 20 μl of VAHTSTM DNA Clean Beads to the supernatant and vortex the mixture. Incubate at room temperature for 5 minutes.
- Spin down the tube and place it on magnets for 3~5 minutes. Discard the supernatant while it is clear.
- Keep the tube on magnets and add 200 μl of newly prepared 80% ethanol. Incubate for 30 seconds then gently discard the supernatant.
- Repeat step 5. To remove most of the liquid in the tube, spin-down is recommended while only small amount of liquid is left. After the mixture is re-separated on magnets, use smaller pipette tips to drip and discard the supernatant remained.
- Air-dry the beads for 5~8 minutes until the beads surface is not reflective without cracking.
- Mix the dried beads with 20 μl of NF-H2O (or TE buffer). Vortex the mixture then placed on the magnets for 3 minutes. Collect the supernatant while it is clear.
-
Drip 1 μl of the size-selection product and measure the concentration via Qubit dsDNA HS Kit.
Detection via Agilent 2100 High Sensitivity Chip is selective.
QC Standards: Production is greater than 300 ng. Main peak locates at around 400~600 bp.
-
| Name | Sequence |
|---|---|
| DNB library oligo1 | TGTGAGCCAAGGAGTTGNAACTGCTGACGTACTGAGAGGCATGGCGACCTTATCAGNNNNNNNNNNNNNNNNNNNNNNNNNTTGTCTTCCTAAGACCG |
| DNB library oligo2 | /5phos/CTTGGCCTCCGACTTAAGTCGGATCGTAGCCATGTCGTTC |
| Splint oligo1 | TCGGAGGCCAAGCGGTCTTAGGAA |
| CID sequencing primer | CTGCTGACGTACTGAGAGGCATGGCGACCTTATCAG |
| DNB Library F_primer | /5phos/TGTGAGCCAAGGAGT |
| DNB Library R_prime | GAACGACATGGCTA |
| Capture probe | CTGCTGACGTACTGAGAGGCATGGCGACCTTATCAGNNNNNNNNNNNNNNNNNNNNNNNNNTTGTCTTCCTAAGACNNNNNNNNNNTTTTTTTTTTTTTTTTTTTTTTTV |
| Stereo-TSO | CTGCTGACGTACTGAGAGGC/rG//rG//iXNA_G/ |
| cDNA PCR primer | CTGCTGACGTACTGAGAGGC |
| Stereo-seq Library F-primer | /5phos/CTGCTGACGTACTGAGAGG*C*A |
| Stereo-seq Library R-primer | GAGACGTTCTCGACTCAGCAGA |
| Stereo-seq DNB splint oligo | GTACGTCAGCAGGAGACGTTCTCG |
| Stereo-seq sequencing primer1 | CTGCTGACGTACTGAGAGGCATGGCGACCTTATCAG |
| Stereo-seq MDA primer | TCTGCTGAGTCGAGAACGTC |
| Stereo-seq sequencing primer2 | GCCATGTCGTTCTGTGAGCCAAGGAGTT |