Tutorial
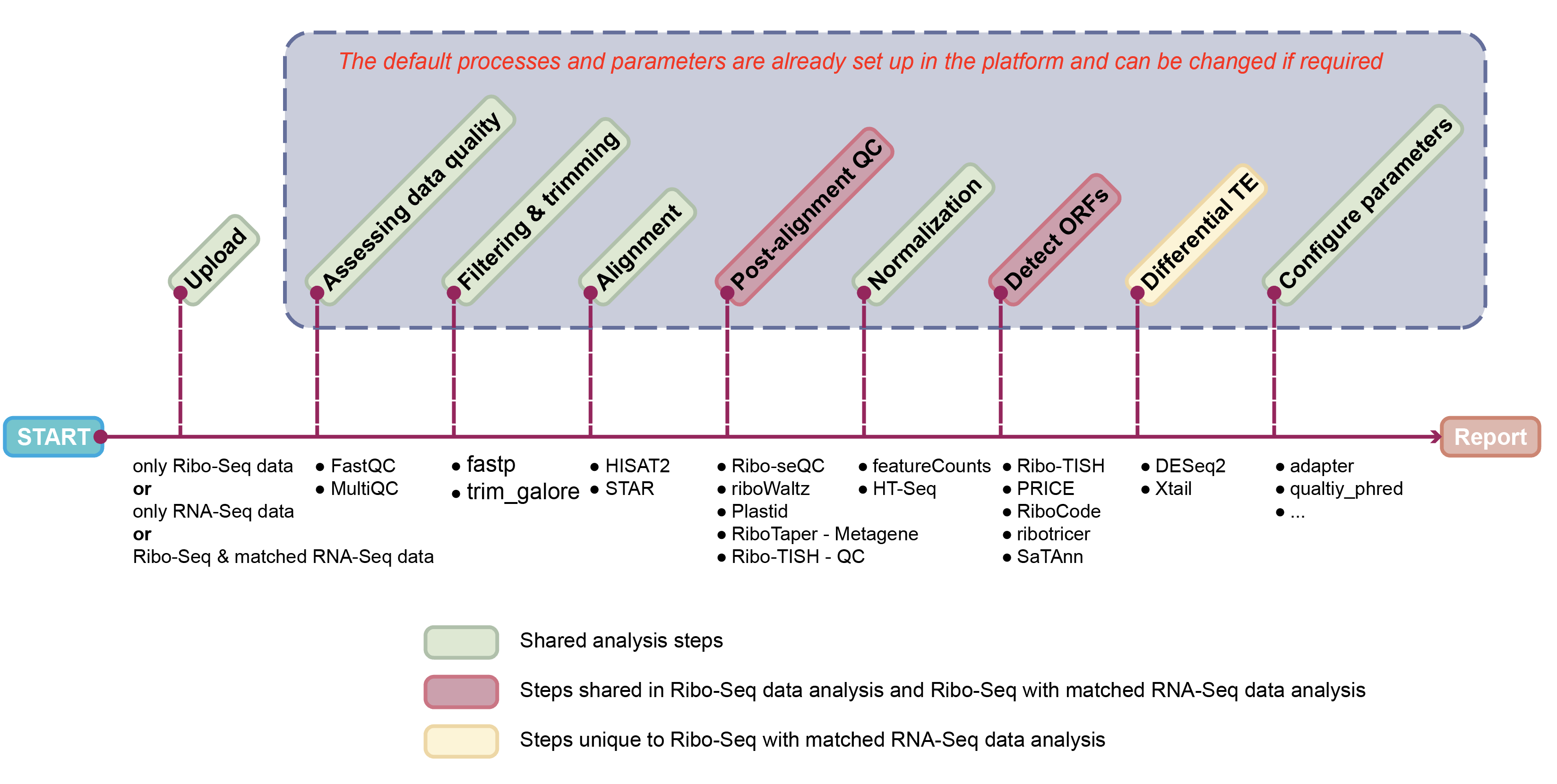
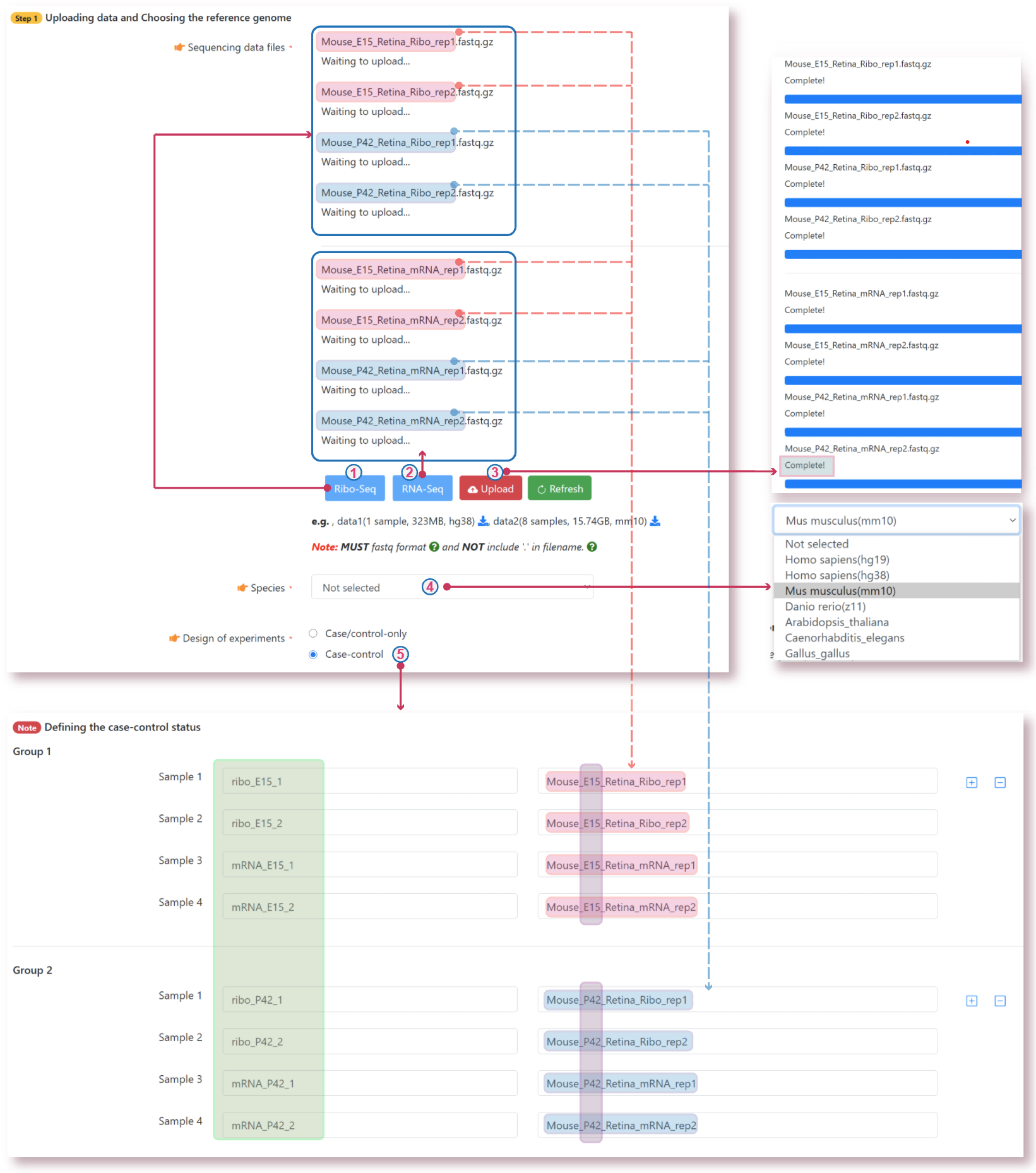
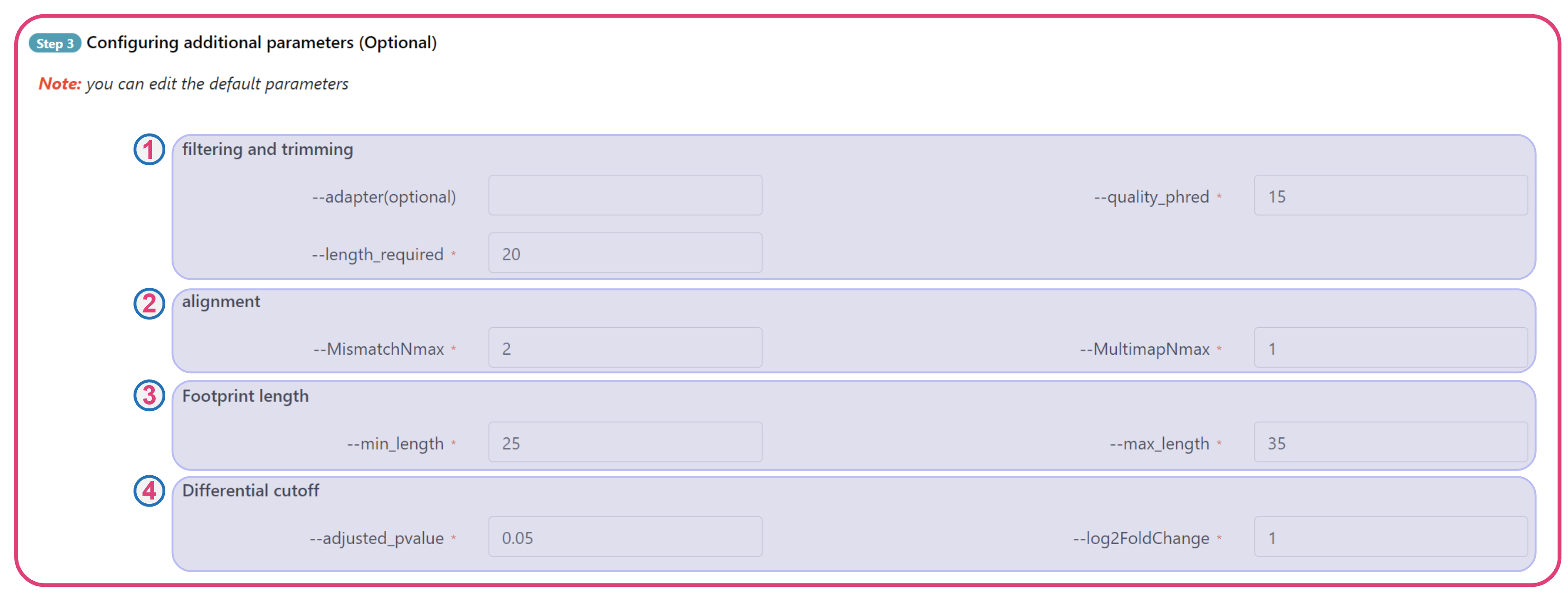
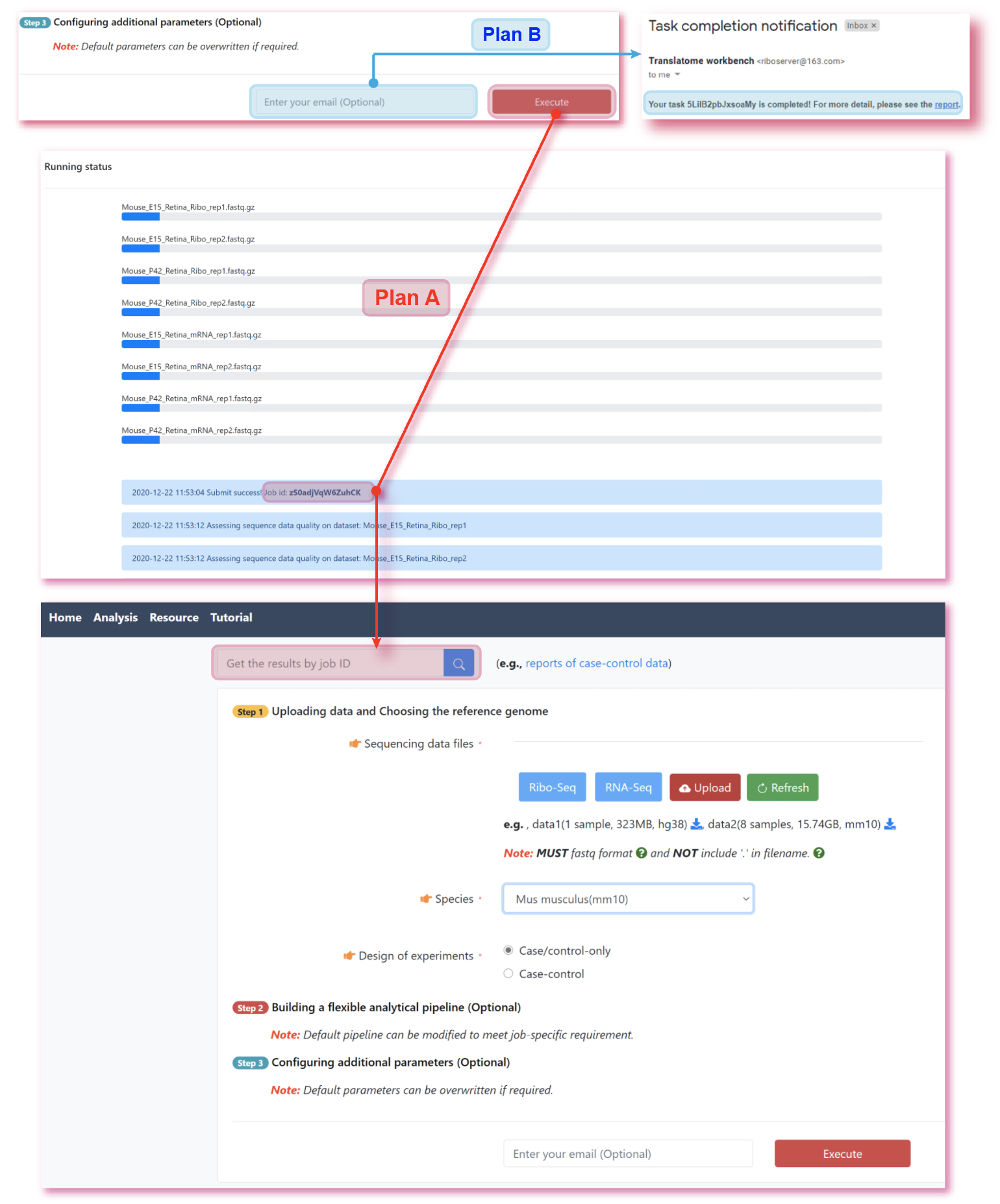
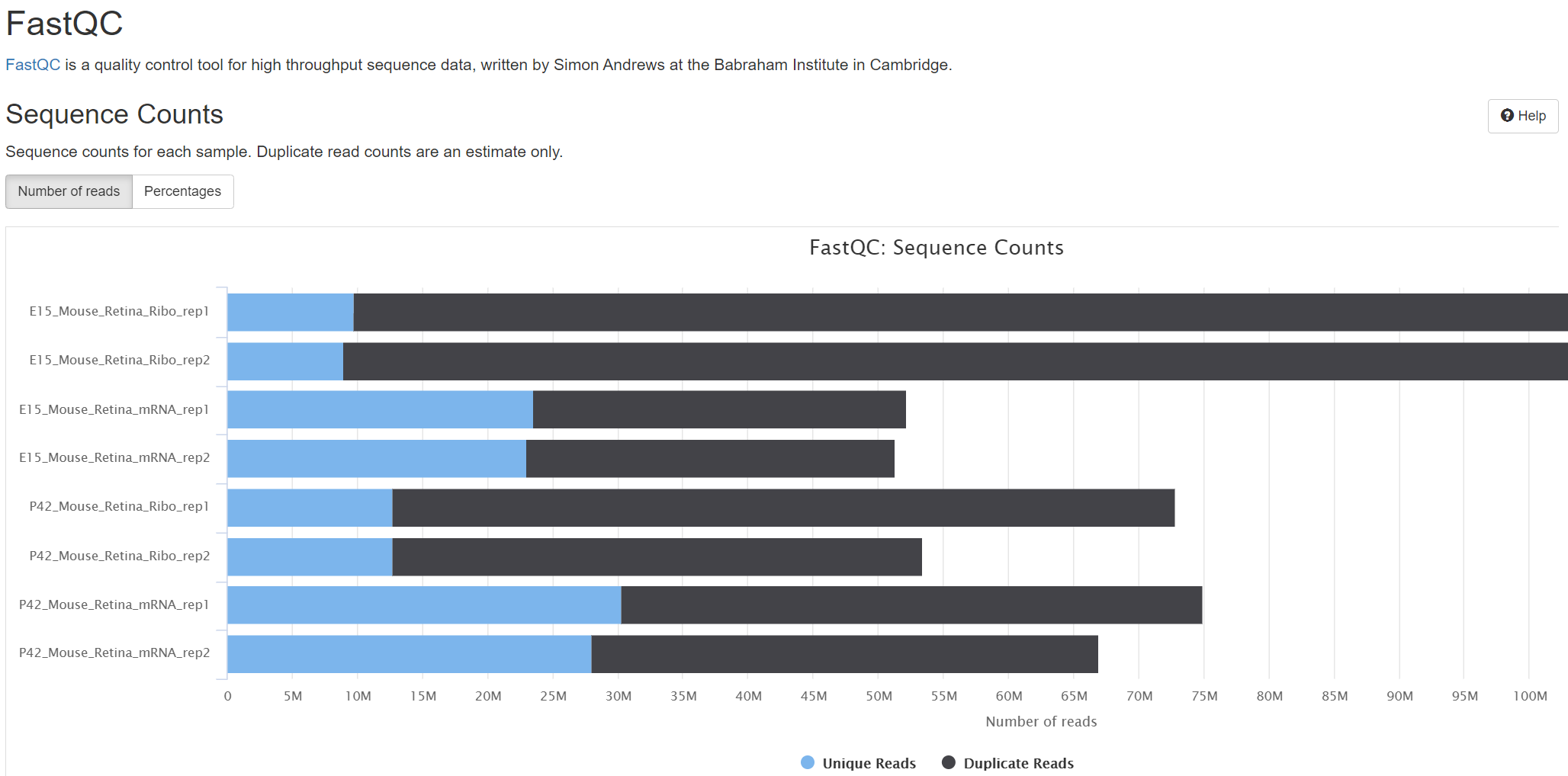
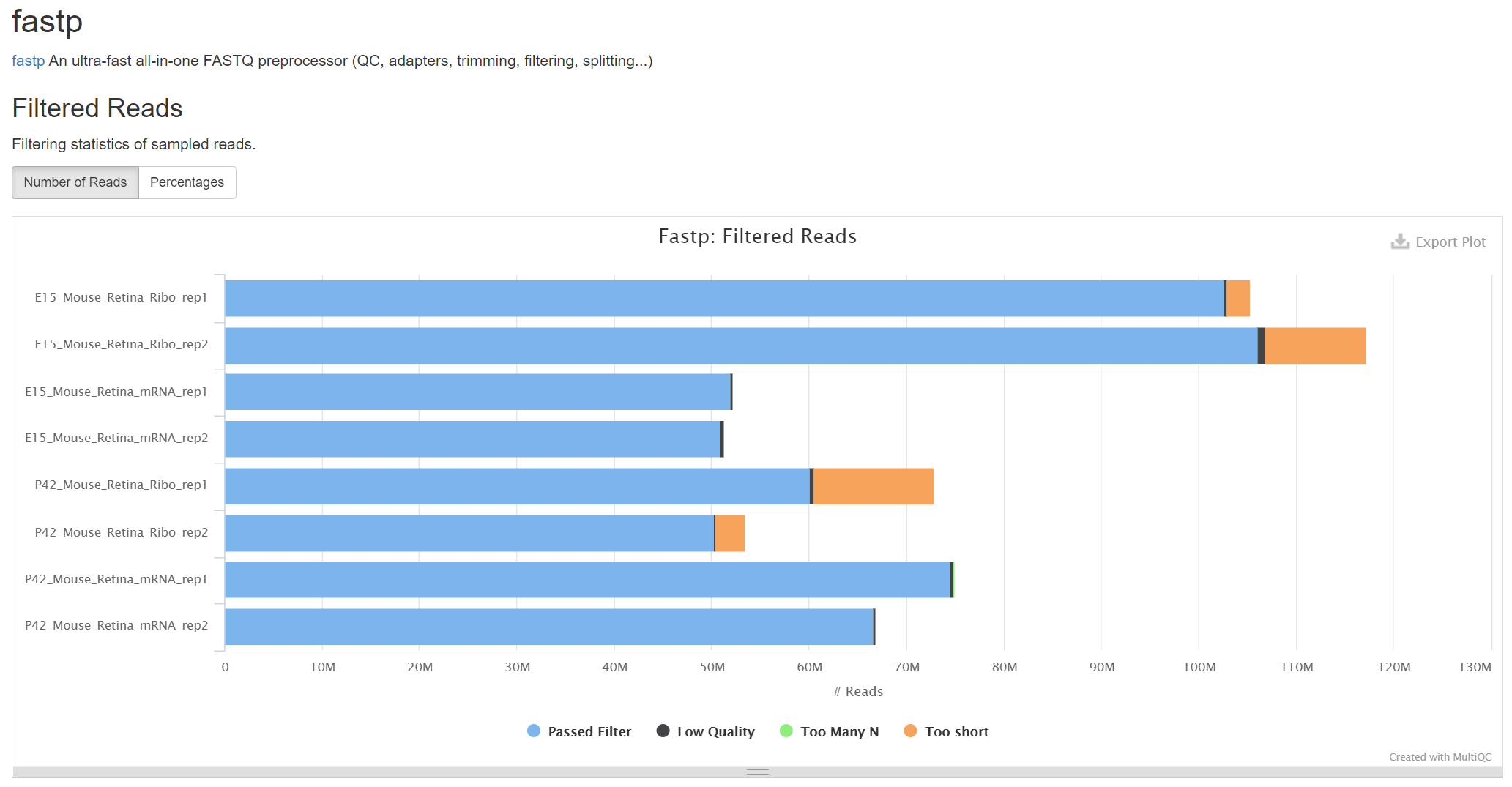
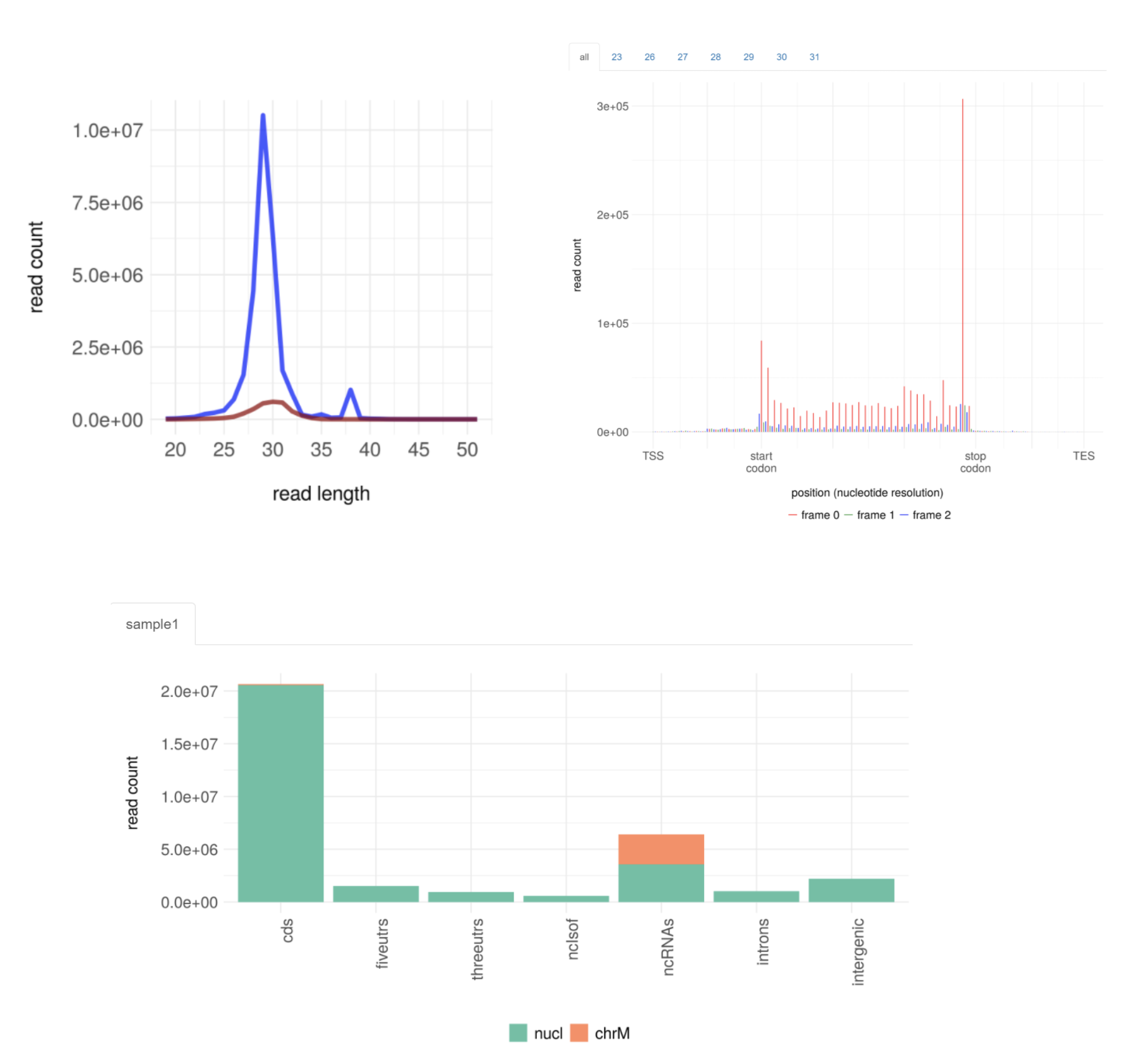
The translatome workbench is a convenient, freely available, web‑based platform to allow effortless analysis of ribosome profiling data in an automatic manner for experienced bioinformaticians, as well for wet‑lab biologists with minimum bioinformatics knowledge. It embeds more than 20 popular analytics tools that provides a full pipeline to cover all key steps for ribo‑seq data analyses, from raw read mapping, filtering and normalization, to the computation of translation‑associated contents such as offset estimation, triplet periodicity, actively translated ORFs detection, and finally to the visualization of analysis results, along with a document report. Additionally, considering that Ribo‑seq and RNA‑seq are often sequenced in parallel, it can also be used to carry out the processing of RNA‑seq data and the integrated analysis of Ribo‑seq and RNA‑seq data, such as differential translational efficiency analysis. Overall, this web server will bring an unprecedented level of convenience for the researcher to decipher information embedded within Ribo‑seq data.